
The Stokes shift
An important trait of fluorescence is the Stokes shift. It describes the differences in the energy level of an exciting and an emitted photon. The photon emitted by a fluorochrome is of greater wavelength than an exciting photon. This is due to the release of energy to the surroundings after the fluorochrome is excited but before it emits the photon. The resulting shift in wavelength makes it possible to distinguish between the exciting and the emission light. The absorption and emission of energy can be seen as specific characteristics of a molecule species.
A fluorescence microscope
A fluorescence microscope (upright or inverted) is similar to an ordinary light microscope, except that the illumination is provided by a laser as monochromatic light or a bright and powerful light source like a mercury-vapor or a xenon arc lamp. In addition it contains an excitation filter and an emission filter. The excitation filter transmits only light that is able to excite the specimen with its particular dye. The light emitted by the specimen has to pass through the emission filter before it reaches the detector. The emission filter is only translucent for light with a distinct wavelength, like the light emitted by the specimen.
In recent years, LEDs (light emitting diodes) have also been used as light source for fluorescence microscopes. The wavelength of the light emitted by LEDs depends on the material used for production. However, an excitation filter is needed in most cases, as LEDs often emit light in a rather broad wavelength range.
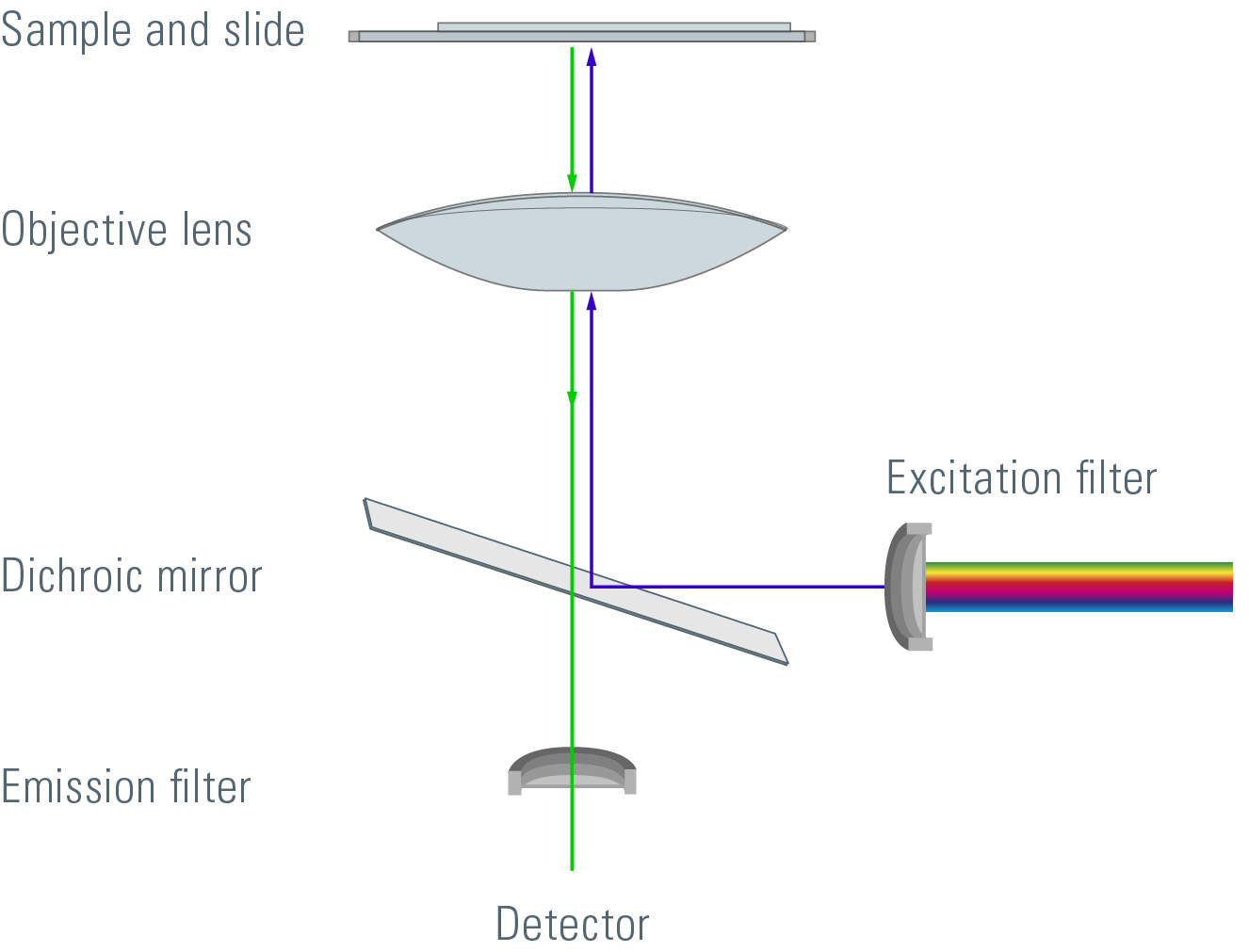
Most fluorescence microscopes are epi-fluorescence microscopes. The illuminator and objective lens are positioned on the same side of the specimen and the light does not pass through the specimen. Besides the excitation and the emission filter, a dichroic mirror is needed for this kind of fluorescence microscope. A dichroic mirror allows light of a certain wavelength to pass through, while light of other wavelengths is reflected. The filters and the dichroic mirror are often plugged in together in a filter cube.
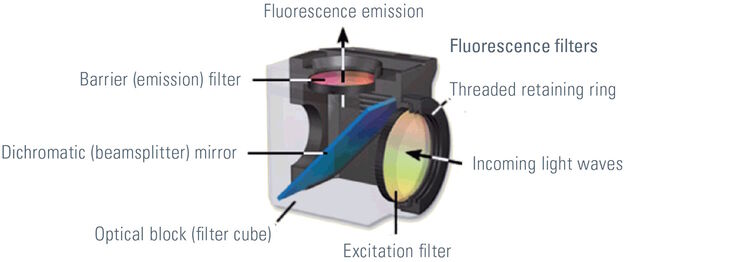
The excitation light passes through the excitation filter and is directed to the dichroic mirror. This reflects the light through the objective towards the specimen. Fluorochromes in the specimen are excited and emit photons. This emission light passes back through the objective to the dichroic mirror. The emitted light has an appropriate wavelength and is able to pass. Excitation light that is reflected by the specimen is not able to pass through the dichroic mirror and will be blocked. If excitation light is able to pass through the dichroic mirror it will be blocked when it reaches the emission filter. Light passing through the emission filter can be measured with a detector.
Different types of filters are used in fluorescence microscopy. Band pass, long pass and short pass filters can be distinguished. Band pass filters transmit a band of wavelengths, whereas light with a greater or smaller wavelength will be blocked. Long pass and short pass filters are edge filters. Long pass filters transmit light of long wavelengths. Light with a wavelength above a certain cutoff value will not be able to pass through. In contrast, short pass filters transmit short wavelengths and block long ones.
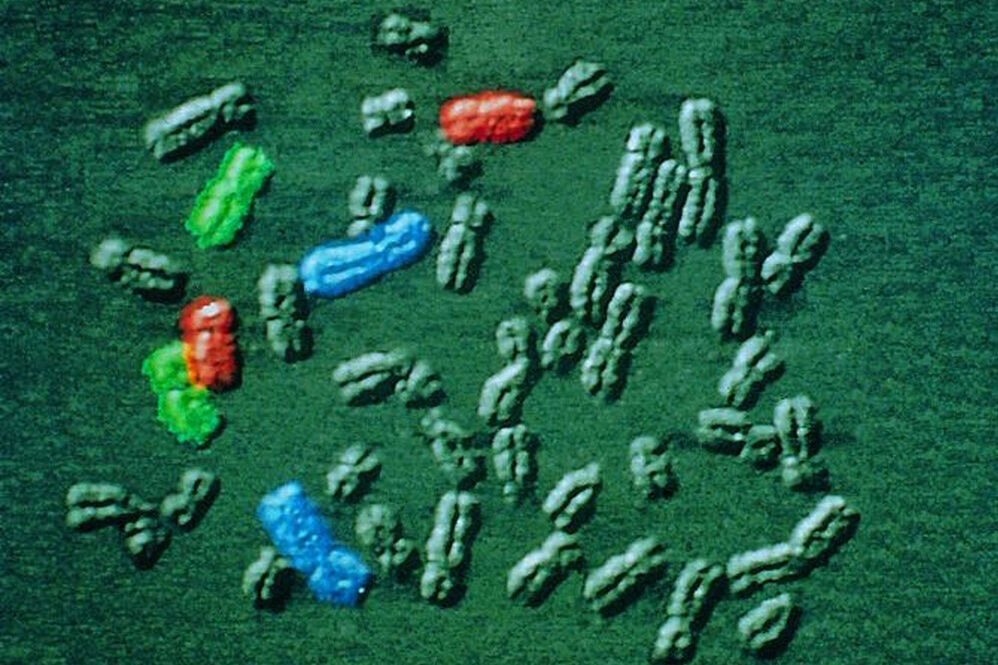
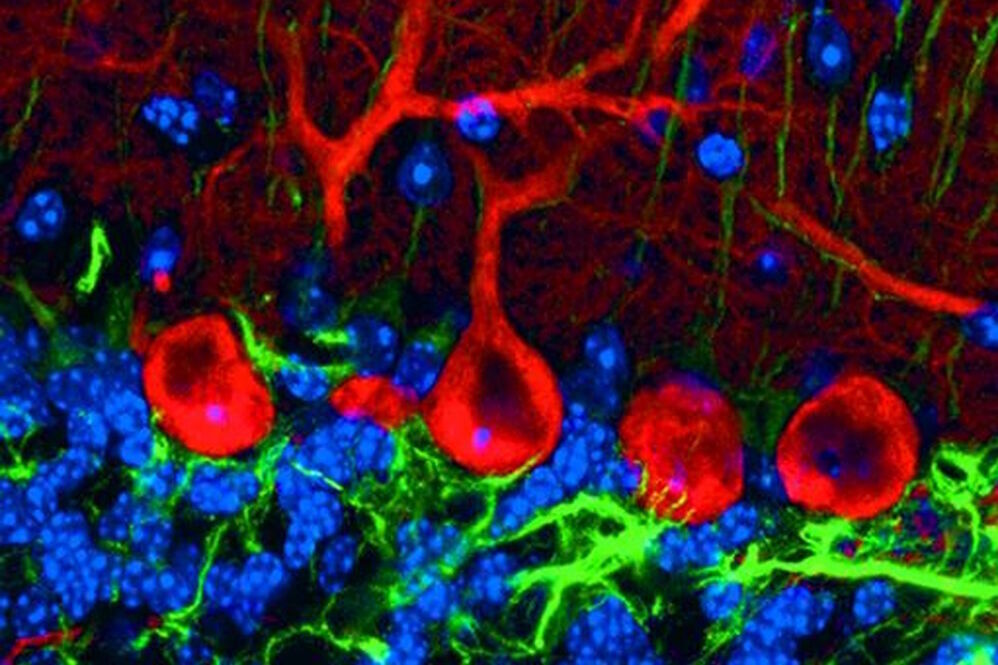
Different techniques in fluorescence microscopy
Fluorescence microscopy is widely used and offers great specificity. Various techniques make it possible to address different problems and even to circumvent the diffraction limit that was described by Ernst Abbe.
The localization of a molecule species can be determined with a co-staining of organelles, e.g. the cytoskeleton or membranes. Confocal laser scanning microscopy (CLSM) makes it possible to observe areas in the specimen without signals from the outside of the focal plane and allows optical sectioning. Total internal reflection (TIRF) microscopy is a technique that allows the observation of a thin region close to the cell surface. An evanescent field excites the fluorochromes in this area.
A technique for observing the dynamics of a molecule species is fluorescence recovery after photobleaching (FRAP). Fluorochromes in a restricted area are photobleached and the diffusion of unbleached molecules into this area can be measured. Interaction studies can be performed with fluorescence energy transfer (FRET) microscopy. An excited donor chromophore can transfer energy to an acceptor fluorochrome and excite it. This is only possible if both are brought together very closely. If these dyes are coupled to different proteins, they will only be able to transfer energy and fluoresce if the proteins interact with each other.
Techniques that allow sub-resolution images are stimulated emission depletion (STED) microscopy, ground-state depletion (GSD) microscopy, single molecule and ground state depletion microscopy followed by individual molecule return (GSDIM), and photoactivated localization microscopy (PALM) and stochastic optical reconstruction microscopy (STORM). The sub-resolution microscopes used for these techniques are also referred to as nanoscopes, as they resolve images at the nanometer scale.
Related Articles
-

Technical Terms for Digital Microscope Cameras and Image Analysis
Learn more about the basic principles behind digital microscope camera technologies, how digital…
Dec 14, 2023Read article -

Epi-Illumination Fluorescence and Reflection-Contrast Microscopy
This article discusses the development of epi-illumination and reflection contrast for fluorescence…
Nov 02, 2023Read article -

Life Science Research: Which Microscope Camera is Right for You?
Deciding which microscope camera best fits your experimental needs can be daunting. This guide…
Jul 31, 2023Read article
Related Pages
-
Fluorescence
Find out how fluorescence microscopes from Leica Microsystems support your research. Fluorescence is…
Visit related page -
How to Prepare your Specimen for Immunofluorescence Microscopy
Immunofluorescence (IF) is a powerful method for visualizing intracellular processes, conditions and…
Visit related page
