Localization microscopy
Localization based super-resolution techniques such as STORM, dSTORM/GSDIM or (spt)PALM are based on temporally decorrelated fluorescence. By achieving isolated molecules’ emission, their subsequent localization with subdiffraction accuracy can be used to reconstruct super-resolved images. In order for these approaches to work, the vast majority of the molecules within the field of view must be in a non-emitting state.
To achieve a temporal separation of fluorescence emissions, several approaches were developed:
- STORM (Stochastic Optical Reconstruction Microscopy) methods achieve a temporally resolved fluorescence of single molecules by putting the majority of the population into a dark state with the help of high power laser illumination [1, 2, 3]. In order for this approach to work the specimen has to be embedded in oxidizing or reducing buffers to stabilize the off state of the fluorophore. Stochastically a subpopulation of the molecules escapes from the dark state and emits fluorescence. This "on rate" can be fine-tuned by the solution surrounding the specimen and illumination with a second laser (405 nm) which reduces the off-state's life time.
- PALM (Photo Activated Localization Microscopy) techniques use photo-activatable/convertible fluorescent proteins that can be "turned" on and off [4, 5]. After activation they are imaged until they bleached. In this case the intensity of the activation light source determines the fluorophores activation rate.
With the help of localization microscopy techniques, a sub-diffraction resolution is achieved and cellular components can be visualized in unprecedented detail. Nevertheless there is a major challenge for those kinds of methods namely attaining super resolved images of native proteins on living cells.
When researchers require to study endogenous proteins at high densities in living cells, neither STORM nor PALM technologies perfectly fulfil all required preconditions. In the case of STORM based approaches the reducing imaging buffers are incompatible with living cells. PALM in turn depends on the utilization of genetically modified chimeric photo-activatable fluorescent proteins, thus endogenous proteins cannot be observed.
Universal Point Accumulation for Imaging in Nanoscale Topography (uPAINT) is a sophisticated technique circumventing these issues [6]. It works by dynamically imaging single-molecule tracks on the cell surface under oblique illumination, hence resulting in a super resolved tracking of native biomolecule behavior at the surface of a living cell.
In contrast to traditional localization microscopy methods, which utilize stochastic fluorescent emission for imaging a small subset of molecules, uPAINT achieves a temporal separation of fluorescence signals by imaging low concentrations of fluorescent ligands as they bind to and interact with their targets inside a small light volume.
The uPAINT principle
Universal PAINT goes back to the original approach called PAINT (Point Accumulation for Imaging in Nanoscale Topography). For this purpose fluorescently labelled probes diffuse in a solution and start to fluoresce upon binding to the object of interest. Alexey Sharanov and Robin Hochstrasser introduced this method by continuously targeting lipid bilayers and large unilamellar vesicles with Nile Red which is non-fluorescent in aqueous solution but starts fluorescing in a hydrophobic environment [7]. Fast molecule diffusion in water leads to a constant flux of Nile Red at the proximity of the membranes. Subsequently a fluorescent signal appears from the moment it enters the membrane. In this new environment the probe becomes fluorescent and its mobility is drastically decreased, allowing its detection with a camera (for several milliseconds) until photo bleaching occurs. By determining the centroid of each single fluorescent signal, a resolution of approx. 25 nm is possible. Nevertheless, owing to the very short insertion duration (milliseconds) it is not possible to track the behavior of single molecules over long time periods.
Later on Gregory Giannone and Eric Hosy focused on and overcame some of the limitations of the original PAINT technique. By using specific fluorescent ligands (i.e. antibodies) instead of nonspecific membrane dyes, dynamic imaging of single, specific protein interactions became possible. Owing to the broader application bandwidth, this method was christened universal PAINT [6].
For this technique background fluorescence of unbound tagged ligand present in the medium is overcome by using oblique illumination (Figure 1). Diverting a TIRF laser from its intended use is the key. Sending the laser slightly below the critical angle to the glass slide leads to the formation of a "Highly Inclined and Laminated Optical sheet" (HILO) passing obliquely through the specimen. Only fluorophores within this volume are excited.
Low concentrations of labelled ligands are added to the buffer inside the medium bathing the specimen. Brownian diffusion leads to the continuously and stochastically binding of ligands to their targets at the cell surface. Only fluorophores within the HILO beam path are excited and imaged (Figure 1).
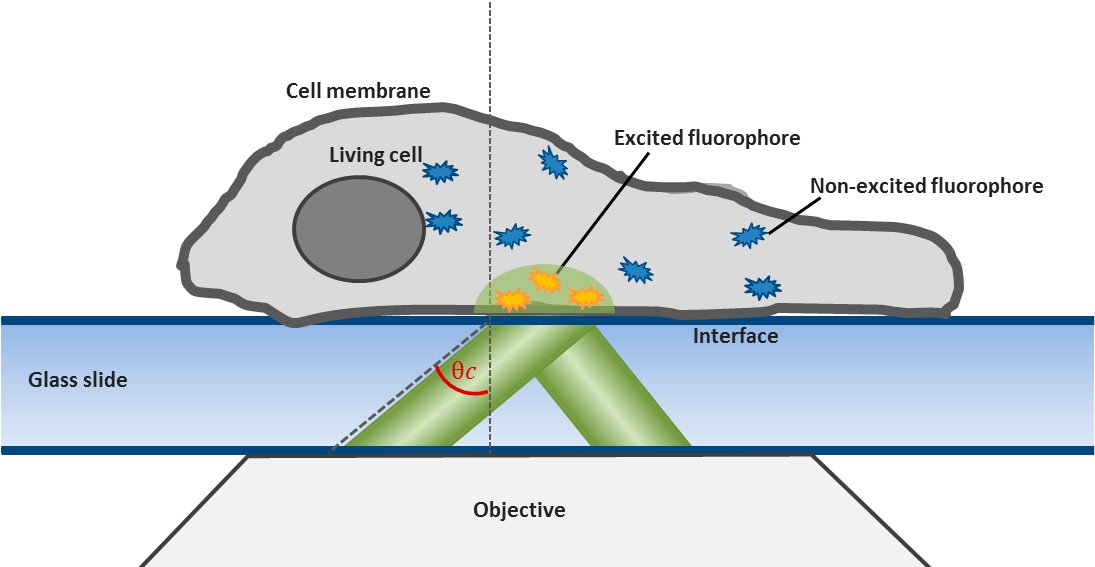
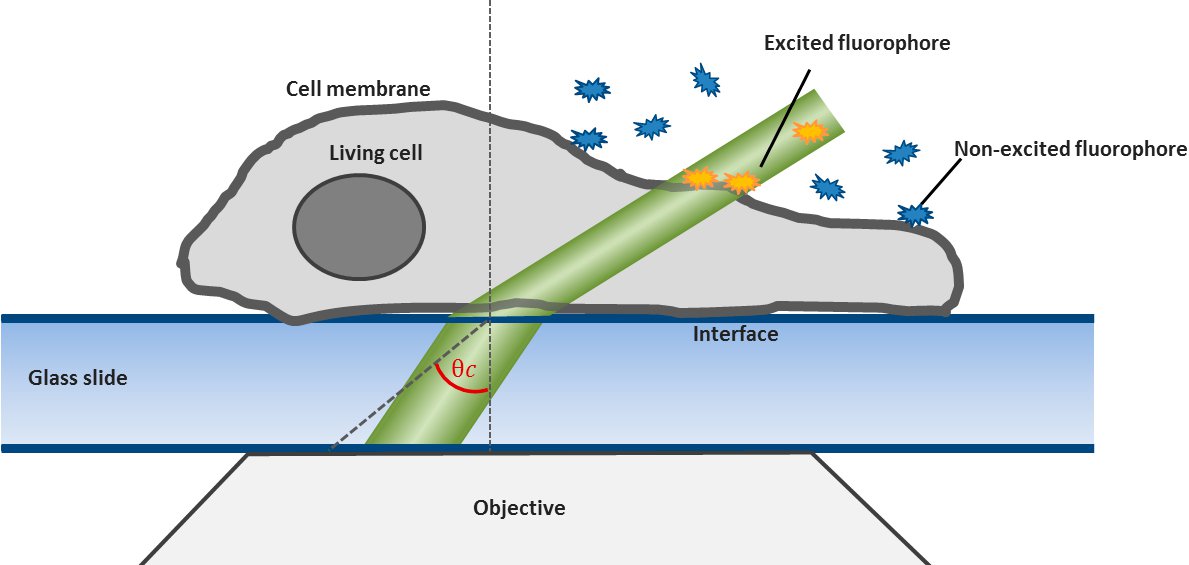
Fig. 1: The uPAINT principle. A) The Leica SR GSD 3D offers the opportunity to adjust the penetration angle of the laser beam. With it TIRF (Total Internal Reflection Fluorescence) illumination can be achieved, if the laser hits the glass slide-specimen interphase beyond the so called critical angle θ
In contrast to the relatively short and insertion dependent fluorescence of lipid probes used for PAINT, the signals from ligands used for uPAINT is predominantly limited by photobleaching of the dye rather than ligand dissociation rates.
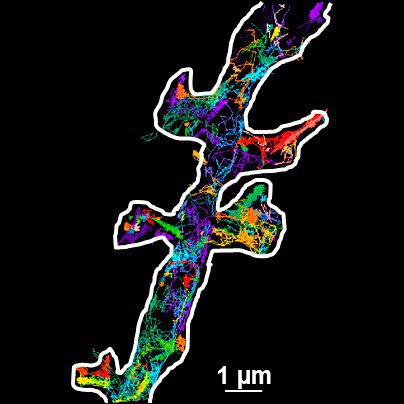
Thus the bound ligand's motion can be tracked and recorded with sub diffraction resolution. Subsequent data analysis and reconstruction of the ligands movement results in so called trajectories, indicating their routes. Due to the huge amount of data which can be gathered, statistics concerning the ligands' speed, confinement, cluster organization etc. can be extracted, for example to study membrane receptors at synapses (Figure 2).
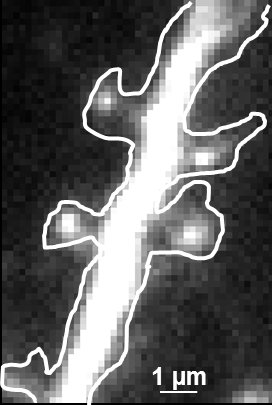
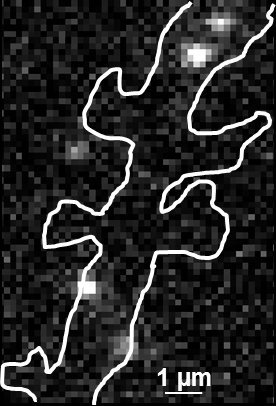
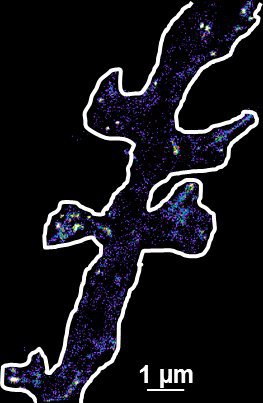

Fig. 2: The different steps of uPAINT image formation. A) Epifluorescence detail of a neuron transfected with the synaptic marker Homer-GFP (to outline the cell). B) Example of a single image acquired during the experiment. Four well isolated single fluorescing ligands (ATTO 647) bound to their target structure (not specified). Each individual particle will be super-localized by computing (pixel size 160 nm). C) Over-accumulation of all the single particle localizations. Bright pixels correspond to highly visited areas of the dendrite (pixel size 20 nm). D) Trajectories of individual proteins. Each color corresponds to the journey of a single particle. Super-resolution images have been obtained using a software developed in JB Sibarita's team (Bordeaux University), commercially available as WaveTracer MetaMorph plugin.
Various biological questions can be explored with uPAINT. Thereby, the respective fluorescent probes have to be chosen carefully according to different purposes. For example to reach restricted areas, small probes such as TrisNTA-atto or Fab fragment can be used (around 2 nm [6]). To record long trajectories, multi-coupled antibodies are preferable. Interestingly even single molecule FRET experiments can be developed [8].
In summary uPAINT not only generates super resolved images but also provides super resolved dynamic information on single, specific, membrane bound biomolecules in living cells. In contrast to classical localization microscopy techniques, uPAINT circumvents the usage of harmful buffers or genetically modified proteins and accordingly enables observation under near natural conditions [9].
The one limitation of the uPAINT technique is that it is restricted to the studies of plasma membrane bound molecules. Since fluorescently labelled antibodies, cannot penetrate the intact plasma membrane of a living cell, it is not possible to use uPAINT to study intracellular protein dynamics.
uPAINT with the Leica SR GSD 3D
The Leica SR GSD 3D offers two basic operation modes: Epifluorescence and TIRF illumination. For TIRF (Total Internal Reflection Fluorescence) illumination the TIRF illuminator collimates a laser beam to the cover glass, containing the specimen in aqueous solution. If the laser beam exceeds a critical angle to the glass slide-water-interphase, total reflection occurs. At the same time an evanescent wave spreads into the specimen at the other side of the glass slide. Its penetration depth can be manipulated from 80 to 250 nm by tilting the laser angle. Only fluorophores inside the evanescent field will be excited, resulting in a very good signal to noise ratio. For this reason TIRF illumination is a preferred technique to observe processes near the plasma membrane (Figure 1).
By shifting the laser incidence below the critical angle, it passes the glass slide and subsequently illuminates the specimen obliquely. Means only a small portion of the sample is illuminated by the excitation light. Images acquired in this so-called HILO (Highly Inclined and Laminated Optical sheet) mode show a low background and good signal to noise ratio. Furthermore bleaching and phototoxicity can be reduced with this method resulting in prolonged cell viability (Figure 1).
In practice the Leica SR GSD 3D offers two different working modes to operate the laser beam for TIRF respectively HILO illumination (see Figure 3).
The Automatic Mode allows choosing predefined penetration depths stepwise between 80 and 250 nm. Furthermore the direction of the evanescent field (azimuth) can be determined in four different ways.
The Expert Mode allows free selection of the penetration depth of the evanescent field by moving the slider within the green area (see Figure 3). Beyond the green zone the laser exceeds the critical angle and starts to induce HILO oblique illumination.
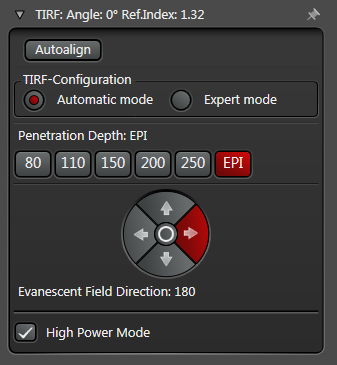
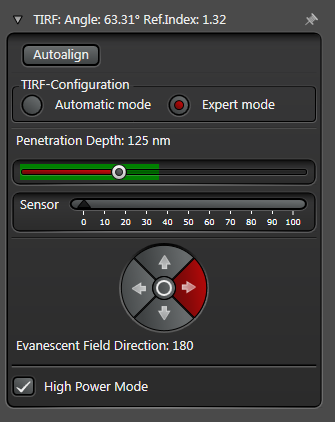
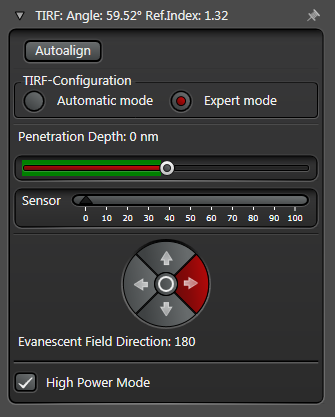
Figure 3: Automatic vs. Expert Mode: Penetration depth and azimuth of the excitation laser beam can be controlled by the imaging software. A) The Automatic mode enables to choose the penetration depth of the evanescent field stepwise from 80 to 250 nm plus four different evanescent field directions. B) The Expert mode allows setting the penetration depth step-less with a slider. The green area marks the TIRF zone. C) Putting the slider out of the green TIRF zone achieves HILO illumination.
For a uPAINT experiment, cells grown on glass bottom chambers should be covered with a non-fluorescent medium (ionic solution such as Tyrode’s medium) and placed on the microscope. After searching the cells in the brightfield mode one can identify and focus on an interesting area for observation. Subsequently the microscope has to be switched to the GSD mode, the correct filter cube and laser has to be chosen. Now the Expert Mode helps to find the correct settings for HILO illumination (see above). Note that the position of the slider and in turn the penetration angle of the laser depends on the thickness of the cells. Afterwards fluorophore-coupled antibodies or ligands have to be added to the medium. During subsequent acquisition only fluorophores bound to the relevant antigen at the plasma membrane will fluoresce. Owing to the oblique illumination, unbound ligands will not be excited, preventing their bleaching and decreasing the noise level. In the course of the experiment the constant labelling of the plasma membrane proteins can be imaged. Raw data files can subsequently be used to reconstruct localization and dynamics of the single molecules in super-resolution.
uPAINT experiments can sometimes require long acquisition periods (tens of seconds) to reveal the movement and organization of almost all proteins of interest. In those circumstances, drift can be corrected by image to image detection of a bead's position (f.e. TetraSpeckTM). After acquisition, the Leica SR GSD 3D software is able to automatically reposition each image by the bead's localization. This drift correction mode is essential when studying nanoscale clusters for example.
To conclude, the Leica SR GSD 3D offers different illumination options. Its original use is to do dSTORM/GSDIM super-resolution microscopy under TIRF or epifluorescence illumination. In addition, HILO illumination can be conducted, necessary for uPAINT. Thus both, static as well as dynamic data of proteins can be gathered with one microscope enabling super-resolved examination of the organization plus the dynamics of a protein at endogenous levels on a living cell.
References
- Rust MJ, Bates M, and Zhuang X: Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy (STORM). Nat. Methods 3 (10): 793–96 (2006).
- Fölling J, Bossi M, Bock H, Medda R, Wurm CA, Hein B, Jakobs S, Eggeling C, and Hell SW: Fluorescence nanoscopy by ground-state depletion and single-molecule return. Nat. Methods 5 (11): 943–45 (2008).
- Heilemann M, van de Linde S, Schüttpelz M, Kasper R, Seefeldt B, Mukherjee A, Tinnefeld P, and Sauer M: Subdiffraction-resolution fluorescence imaging with conventional fluorescent probes. Angew. Chem. Int. Ed. Engl. 47 (33): 6172–76 (2008).
- Betzig E, Patterson GH, Sougrat R, Lindwasser OW, Olenych S, Bonifacino JS, Davidson MW, Lippincott-Schwartz J, and Hess HF: Imaging intracellular fluorescent proteins at nanometer resolution. Science 313 (5793): 1642–45 (2006).
- Manley S, Gillette JM, Patterson GH, Shroff H, Hess HF, Betzig E, Lippincott-and Schwartz J: High-density mapping of single-molecule trajectories with photoactivated localization microscopy. Nat. Methods 5 (2): 155–57 (2008).
- Giannone G, Hosy E, Levet F, Constals A, Schulze K, Sobolevsky AI, Rosconi MP, Gouaux E, Tampé R, Choquet D, and Cognet L: Dynamic superresolution imaging of endogenous proteins on living cells at ultra-high density. Biophys. J. 99 (4): 1303–10 (2010).
- Sharonov A, and Hochstrasser R: Wide-field subdiffraction imaging by accumulated binding of diffusing probes. PNAS 50: 18911–16 (2006).
- Winckler P, Lartigue L, Giannone G, De Giorgi F, Ichas F, Sibarita JB, Lounis B, and Cognet L: Identification and super-resolution imaging of ligand-activated receptor dimers in live cells. Sci. Rep. (2013).
- Zimmermann T, Hosy E: Webinar: New Dimensions in Super-Resolution Microscopy. Leica Science Lab (2014).
Related Articles
-
Coherent Raman Scattering Microscopy Publication List
CRS (Coherent Raman Scattering) microscopy is an umbrella term for label-free methods that image…
May 26, 2025Read article -

Mica: A Game-changer for Collaborative Research at Imperial College London
This interview highlights the transformative impact of Mica at Imperial College London. Scientists…
Feb 10, 2025Read article -

How to Study Gene Regulatory Networks in Embryonic Development
Join Dr. Andrea Boni by attending this on-demand webinar to explore how light-sheet microscopy…
Nov 20, 2024Read article
