
IF-Präparate können mit verschiedenen Mikroskopietechniken (z. B. CLSM, Epifluoreszenz, TIRF, GSDIM) analysiert werden, je nach Anwendung oder Interesse des Forschers. Inzwischen ist die IF für eine große Anzahl von Forschungsgruppen, die zumindest Zugang zu einem einfachen Fluoreszenzmikroskop haben, unverzichtbar geworden.
Das Kernelement eines IF-Experiments ist eine Kombination aus zwei verschiedenen Komponenten:
- Zunächst handelt es sich um spezifische Antikörper, die zur Bildung eines Immunkomplexes verwendet werden, um die gewünschten Moleküle - in den meisten Fällen Proteine - in der Zelle zu markieren.
- Zweitens: Fluorochrome, die an die Immunkomplexe gekoppelt sind und so die Zielstrukturen bei der Mikroskopie sichtbar machen.
Typischer Arbeitsablauf
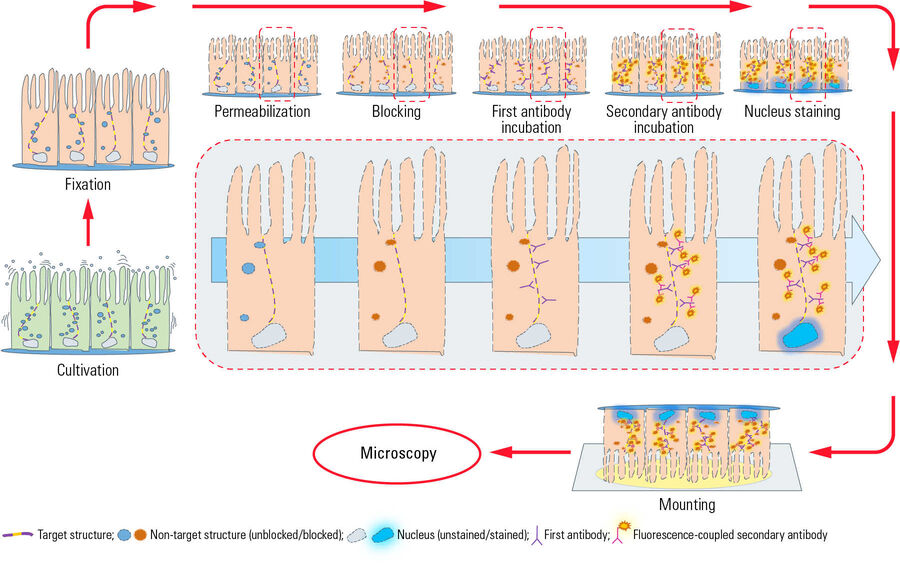
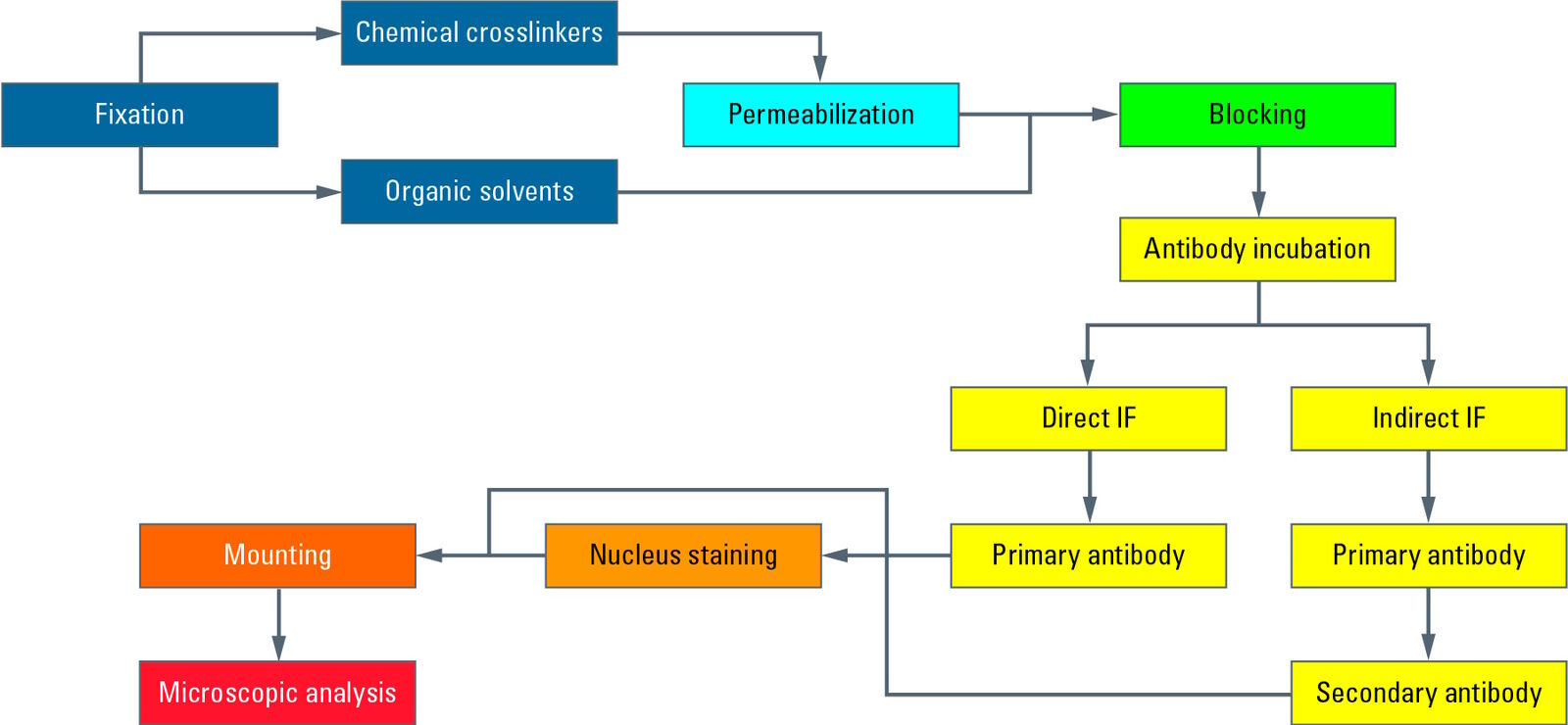
Dieses Diagramm zeigt einen typischen Arbeitsablauf der indirekten Immunfluoreszenz (IF) mit Epithelzellen, die auf einem Deckglas adhärent wachsen. Nach der Kultivierung werden die Zellen mit einem chemischen Vernetzer (z. B. Formaldehyd) fixiert und damit abgetötet. Anschließend erfolgt ein Prozess der Permeabilisierung mit Detergenzien, damit die Antikörper die Zellmembranen passieren können. Die Blockierung mit normalem Serum, Milchpulver oder Rinderserumalbumin reduziert die unspezifische Bindung von Antikörpern an Nicht-Zielstrukturen, um falsch-positive Signale zu minimieren. Anschließend erfolgt die Inkubation mit dem ersten Antikörper, der spezifisch Epitope auf dem Zielmolekül erkennt. In einem zweiten Inkubationsschritt wird der fluoreszenzgekoppelte Sekundärantikörper aufgetragen, der an den ersten Antikörper bindet und somit die Zielstruktur sichtbar macht. Nach der Antikörperinkubation werden die Zellkerne mit Farbstoffen wie DAPI oder Hoechst gefärbt, die sich in die DNA einlagern. Nachdem das Deckglas mit einem Einbettungsmedium (z. B. Mowiol oder Prolong Gold) auf einem Objektträger befestigt wurde, ist das IF-Präparat für die Mikroskopie bereit.
Direkte vs. indirekte Immunfluoreszenz
Je nach Art des Experiments gibt es zwei verschiedene Varianten der Immunfluoreszenz (IF): Bei der direkten oder primären Immunfluoreszenz (IF) wird ein spezifischer primärer Antikörper, der mit einem Fluorochrom verbunden ist, für die Bindung an die Zielstruktur und deren direkte Visualisierung verwendet.
Für die zweite Variante, die als indirekte oder sekundäre Immunfluoreszenz bezeichnet wird, wird eine zweistufige Inkubation durchgeführt. Zunächst erkennt ein spezifischer primärer Antikörper die Zielstruktur. Danach wird ein mit Fluorochrom gekoppelter sekundärer Antikörper aufgetragen, der sich spezifisch an den primären Antikörper bindet. Diese Spezifität wird erreicht, indem der sekundäre Antikörper gegen die Spezies gerichtet wird, gegen die der primäre Antikörper gezüchtet wurde (siehe Antikörper und Fluorochrome). Vergleicht man die beiden Arten der Immunfluoreszenz, so weist jede von ihnen unterschiedliche Vor- und Nachteile auf:
Durch die Kopplung des primären Antikörpers mit einem Fluorochrom ist die direkte Immunfluoreszenz (IF) schneller als die indirekte Variante, da zeitaufwändige Wasch- und Inkubationsschritte entfallen. Daher ist die direkte Immunfluoreszenz (IF) einfacher zu handhaben und eignet sich besser für die schnelle Analyse von Proben in standardisierten Immunfluoreszenz (IF)-Experimenten, zum Beispiel in der klinischen Praxis. Allerdings ist es unerlässlich, einen gut funktionierenden primären Antikörper mit hoher Affinität zu seinem Antigen zu verwenden. Dies ist gleichzeitig ein negativer Aspekt, denn fluoreszenzgekoppelte und validierte Primärantikörper sind teuer. Außerdem benötigt man für jede Zielstruktur einen eigenen primären Antikörper, und die Verknüpfung des Antikörpers mit einem Fluorochrom bei der direkten Immunfluoreszenz schränkt die Flexibilität bei der Versuchsplanung im Vergleich zur indirekten Immunfluoreszenz ein.
Diese Flexibilität ist ein wesentlicher Vorteil der indirekten Immunfluoreszenz (IF). In der Regel müssen während einer Immunfluoreszenzreaktion mehrere verschiedene Zielstrukturen in derselben Probe sichtbar gemacht werden, weshalb für jedes Zielmolekül ein eigenes Fluorochrom ausgewählt werden muss. Bei der indirekten Immunfluoreszenz können verschiedene fluoreszenzgekoppelte Sekundärantikörper mit verschiedenen Primärantikörpern kombiniert werden (natürlich unter Berücksichtigung der Speziesreaktivität). Wenn Sie dagegen bei der direkten Immunfluoreszenz mit Farbkombinationen Ihrer Zielstrukturen "spielen" wollen, benötigen Sie für jede Farbe einen eigenen Primärantikörper. Ein weiterer Vorteil der indirekten Methode ist die Signalverstärkung durch den sekundären Antikörper. Mehrere sekundäre Antikörpermoleküle können an einen primären Antikörper binden, was zu einer Erhöhung der Fluoreszenz führt, so dass weniger primärer Antikörper aufgetragen werden muss.
Der Arbeitsablauf der indirekten Immunfluoreszenz (IF) nimmt zwar mehr Zeit in Anspruch, ist aber aufgrund der möglichen Kombinationen von primären und sekundären Antikörpern und eines allgemein kostengünstigeren Verfahrens für die meisten Forscher die bevorzugte Option.
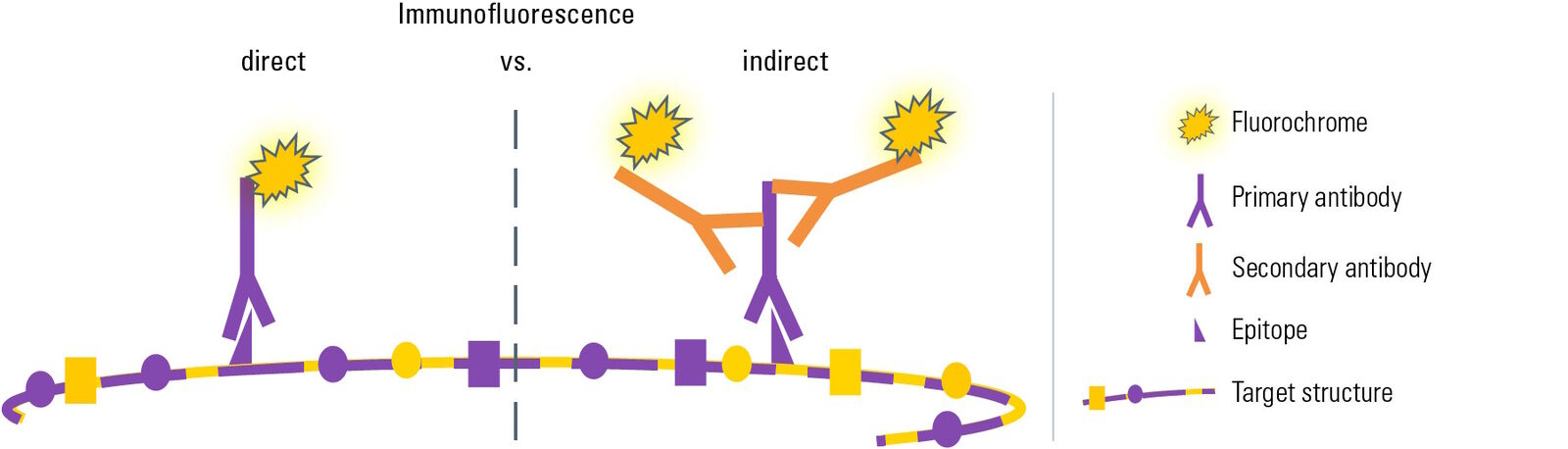
Abb. 2: Eine Zielstruktur kann auf zwei Arten durch Immunfluoreszenz sichtbar gemacht werden: Bei beiden Arten wird ein spezifischer erster Antikörper verwendet, der ein bestimmtes Epitop auf dem Zielmolekül erkennt. Hier besteht das Zielmolekül aus mehreren identischen Proteinuntereinheiten (= Makromolekül) und weist daher auf jeder Untereinheit mehrere Epitope desselben Typs auf. Zur Vereinfachung ist hier nur ein Epitop abgebildet
Bei der direkten Immunfluoreszenz (IF) wird der erste Antikörper direkt mit einem Fluorochrom verbunden, das die Zielstruktur unter dem Mikroskop sichtbar macht.
Bei der indirekten Immunfluoreszenz (IF) wird während eines zweiten Inkubationsschritts ein fluoreszenzgekoppelter Sekundärantikörper aufgetragen, der den ersten Antikörper spezifisch markiert. Dies führt zu einer größeren Flexibilität bei der Auswahl von Antikörpern und Fluorochromen und darüber hinaus zu einer Signalverstärkung, da mehrere sekundäre Antikörpermoleküle an einen primären Antikörper binden können.
Antikörper und Fluorochrome
Das wichtigste Hilfsmittel für eine qualitativ hochwertige Immunfluoreszenzfärbung (IF) ist ein guter Primärantikörper. Mittlerweile gibt es für fast jedes Protein in fast jedem Zelltyp einen oder sogar mehrere kommerziell erhältliche Antikörper. Allerdings gibt es hier ein paar besonders wichtige Faktoren zu beachten.
Um auf der Grundlage Ihrer Immunfluoreszenzfärbung eine präzise wissenschaftliche oder klinische Aussage treffen zu können, müssen Sie die Spezifität Ihres primären Antikörpers für sein Zielantigen sicherstellen. Dabei sollten Sie sich nicht ausschließlich auf die Angaben Ihrer kommerziellen Anbieter verlassen. Wählen Sie Ihren Antikörper entsprechend den bereits verwendeten und validierten primären Antikörpern in der Fachliteratur zu diesem Thema. Prüfen Sie das Datenblatt des Antikörpers auf der Website des Herstellers auf verfügbare Bilder von Immunfluoreszenzfärbungen und vergleichen Sie diese mit Ihren Erwartungen oder anderen veröffentlichten Abbildungen. Achten Sie auf die Klonalität des Antikörpers, da monoklonale Antikörper spezifisch nur an ein Epitop binden, während polyklonale Antikörper mehrere Epitope erkennen, was eine unspezifische Markierung von Nicht-Zielstrukturen wahrscheinlicher macht. Daher sind monoklonale Antikörper mit ihrer hohen Spezifität und guten Affinität in der Regel teurer, erzielen aber auch bessere Ergebnisse.
Wenn Sie eine indirekte Mehrfarben-Immunfluoreszenzfärbung durchführen wollen, müssen die verschiedenen primären Antikörper von unterschiedlichen Spezies stammen, damit die Immunkomplexe durch anschließende Markierung mit fluoreszenzgekoppelten sekundären Antikörpern unterschieden werden können (siehe Tabelle 1). Nehmen wir an, Sie führen eine Immunfluoreszenzfärbung mit einem Antikörper gegen Protein A von der Maus, einem Antikörper gegen Protein B vom Kaninchen und einem Antikörper gegen Protein C von der Ratte durch. Bei der Auswahl der sekundären Antikörper müssen Sie bedenken, dass jeder von ihnen spezifisch nur einen primären Antikörper erkennt. Außerdem müssen sich die Fluorochrome in diesem Beispiel der drei sekundären Antikörper in ihrem Wellenlängenspektrum unterscheiden, um ihr Fluoreszenzsignal bei der mikroskopischen Analyse unterscheiden zu können. Heutzutage sind an Fluorochrome gekoppelte sekundäre Antikörper mit einem Wellenlängenbereich von ultraviolett bis infrarot gegen primäre Antikörper von fast allen Spezies erhältlich. Somit ist der Forscher nur durch die Konfiguration der verfügbaren Mikroskope (Filtersätze, Anregungslaser) beschränkt.
 |  |  | |
|---|---|---|---|
| Target protein | Protein A | Protein B | Protein C |
| Target species | Human | Human | Human |
| First antibody | Anti-Protein A | Anti-Protein B | Anti-Protein C |
| First antibody species reactivity | Mouse anti-human | Rabbit anti-human | Rat anti-human |
| Second antibody species reactivity | Goat anti-mouse | Goat anti-rabbit | Goat anti-rat |
| Fluorochrome excitation/emission | 490/525 nm | 556/573 nm | 650/665 nm |
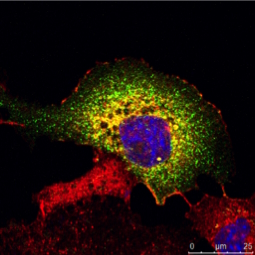
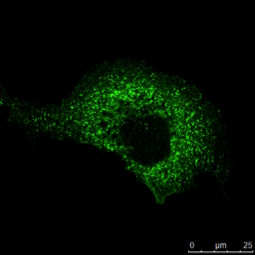
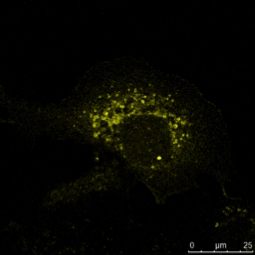
Tab. 1: Dieses Beispiel einer mehrfarbigen indirekten Immunfluoreszenz zeigt, wie drei verschiedene Proteine in derselben Zelle gleichzeitig markiert werden können. Die drei ersten Antikörper müssen von verschiedenen Spezies stammen, damit sie mit drei verschiedenen fluorochromgekoppelten sekundären Antikörpern nachgewiesen werden können. Die Fluorochrome der Sekundärantikörper müssen sich in ihrem Wellenlängenspektrum unterscheiden, um eine eindeutige Analyse im Mikroskop zu ermöglichen. Die im Beispielbild dargestellte Zelle wurde zur Kernfärbung zusätzlich mit Hoechst 33342 behandelt. Die Mikroskopie wurde mit einem Leica TCS SP2 durchgeführt.
Probe
Immunfluoreszenzprotokolle gibt es für eine Vielzahl unterschiedlicher Proben oder Präparate. Die einfachste und am häufigsten verwendete Methode ist die Färbung von kultivierten (eukaryotischen) Zellen aus Zellkulturen. Adhärent wachsende Zellen können auf Deckgläsern, Multiwell-Einsätzen oder direkt auf Kulturschalen mit Glasboden ausgesät und zum gewünschten Zeitpunkt für die Immunfluoreszenz (IF) verwendet werden. Immunfluoreszenz (IF) ist auch mit Suspensionszellen möglich, nachdem die Zellen auf einen Objektträger aufgebracht wurden, z. B. durch Cytospin. In beiden Fällen liegt der Schwerpunkt auf der Analyse intrazellulärer Prozesse oder Strukturen und wird als Immunzytochemie (ICC) bezeichnet.
Bei der Immunhistochemie (IHC) hingegen wird das Vorhandensein von Proteinen oder Molekülen in einem gewebespezifischen Zusammenhang untersucht. Dabei werden ultradünne Schnitte von Organpräparaten (in der Regel eingebettet in Paraffin) verwendet, um z. B. die Expression von Proteinen in einem gesunden Organ im Vergleich zu einem kranken zu untersuchen. Neben der Präparation von Gewebeschnitten ist es auch möglich, IF mit ganzen Organismen durchzuführen, ein Verfahren, das als "whole mount IHC" bezeichnet wird. Zu diesem Zweck werden Embryonen verschiedener Modellorganismen wie Maus, Huhn oder Zebrafisch oder Pflanzenmodellorganismen wie Arabidopsis thaliana verwendet. Bei der Whole Mount IHC ist man durch die Größe des Präparats und die damit verbundene Eindringtiefe der Immunfluoreszenz (IF)-Reagenzien begrenzt. Die einzelnen Inkubationsschritte dauern länger als die Färbung von kultivierten Zellen. Außerdem müssen Mikroskope mit spezieller optischer Ausrüstung für die Analyse großer Proben zur Verfügung stehen.
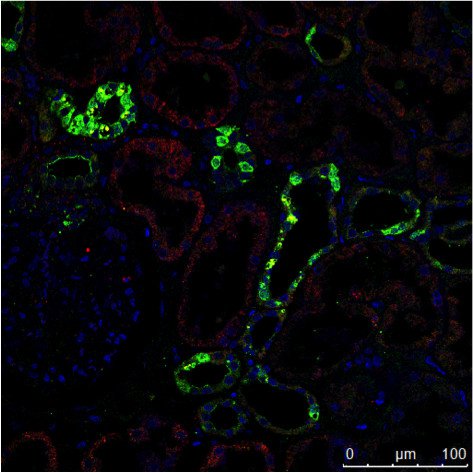
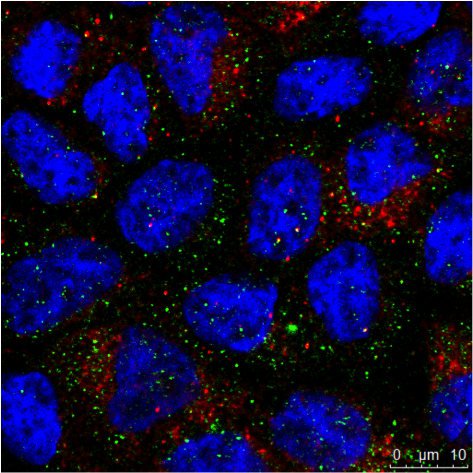
Abb. 3: a) Die immunhistochemische (IHC) Färbung einer menschlichen Niere zeigt verschiedene Zelltypen in verschiedenen Strukturen (z. B. Glomerolus, proximaler Tubulus, distaler Tubulus). Das mit grünem Fluorochrom markierte Protein wird nur von einem bestimmten Zelltyp exprimiert, während das rot markierte Protein weit verbreitet ist. b) Die Aufnahme der Immunzytochemie (ICC) zeigt die Färbung von zwei Proteinen in Zellen desselben Typs (MDCK) durch indirekte Immunfluoreszenz (IF). In diesem Fall ist eine gewebespezifische Untersuchung nicht möglich, sondern die Analyse, ob beide Proteine in der gleichen Struktur kolokalisieren. Für die Kernfärbung wurden beide Proben mit Hoechst 33342 bearbeitet. Die Mikroskopie wurde mit einem Leica TCS SP2 durchgeführt.
Waschschritte
Während eines Immunfluoreszenz-Verfahrens ist besonders auf die Waschschritte zu achten, da die Qualität der Immunfluoreszenz (IF) durch richtiges Waschen verbessert werden kann. Standardmäßig wird PBS als Waschpuffer verwendet, aber auch Varianten wie PBS++ oder PBS-T sind weit verbreitet. PBS++ enthält 1 mM CaCl2 und MgCl2, die vermutlich eine membranstabilisierende Wirkung haben und die Zellablösung verhindern. Zur Erhöhung der Bindungsspezifität der Antikörper wird PBS-T eine Endkonzentration von 0,05 % des Detergens Tween 20 zugesetzt. Es ist äußerst wichtig, den Waschpuffer vorsichtig aufzutragen bzw. wieder abzutrennen, damit sich die Zellen nicht von ihrem Kulturgefäß oder Deckglas lösen. Wenn Sie genügend Zeit haben, sollten Sie zwischen den einzelnen Waschschritten mehrere Minuten einplanen, um eine effiziente Diffusion des Waschpuffers in die Probe zu gewährleisten.
Die einzelnen Waschschritte eines Immunfluoreszenzverfahrens sind im nachstehenden Standardprotokoll aufgeführt.
Fixieren
Das Fixieren ist der erste Schritt in einem Immunfluoreszenz-Verfahren. Ziel ist es, Zellen, Zellverbände oder Gewebe in ihrem aktuellen Zustand zu erhalten und das Mittel durch chemische Substanzen über einen längeren Zeitraum zu konservieren. Während der Fixierung ist es wichtig, dass die zellulären Strukturen so weit wie möglich in ihrer ursprünglichen Form erhalten bleiben. Für die Immunfluoreszenz sind verschiedene Fixierungsmethoden nützlich, wobei jedes Reagenz eine andere Wirkung auf die Epitope des primären Antikörpers hat. Die Bindungsstellen der Antikörper können durch die Fixierung maskiert oder auch beschädigt werden, was die Qualität der Immunfluoreszenzfärbung beeinträchtigt. Da jeder Antikörper in Abhängigkeit von den verschiedenen Fixierungsmitteln unterschiedlich an sein Antigen bindet, ist es notwendig, für einen neuen Antikörper mehrere Fixierungsmethoden auszuprobieren. Häufig finden sich Angaben zu einem geeigneten Fixiermittel auf dem Datenblatt des Antikörpers. Die ideale Fixierung erhält die zelluläre und subzelluläre Architektur und sorgt für unblockierte Antigene, die eine gute Antikörperbindung ermöglichen. In der Praxis muss man ein Gleichgewicht zwischen diesen beiden Faktoren finden.
Generell lassen sich die Reagenzien in zwei Gruppen einteilen: chemische Vernetzer und organische Lösungsmittel.
- Chemische Vernetzer wie Formaldehyd vernetzen die Proteine über ihre freien Aminogruppen; die zelluläre Morphologie bleibt in den meisten Fällen gut erhalten. Allerdings werden auch die Antigene vernetzt, was die Antikörperbindung verringern kann. Glutaraldehyd hat ebenfalls eine konservierende Wirkung auf zelluläre Strukturen, doch führt es bei der Mikroskopie zu einer starken Autofluoreszenz des Präparats (siehe Kontrollen).
- Organische Lösungsmittel wie Methanol oder Aceton haben eine dehydrierende Wirkung und fällen Proteine aus, wodurch sie in ihrem zellulären Kontext fixiert werden. Dabei gehen jedoch lösliche Moleküle und viele Lipidbestandteile verloren. Häufig wird eine Kombination aus Methanol und Aceton verwendet, denn Methanol eignet sich zwar am besten für die Erhaltung der zellulären Strukturen, wirkt sich aber äußerst negativ auf viele Epitope aus. Aceton ist hier weniger schädlich. Sie sollten auch bedenken, dass fluoreszierende Proteine wie GFP, die bereits in Ihren Zellen vorhanden sind, durch die Fixierung mit organischen Lösungsmitteln weitgehend zerstört werden. Wenn der Hersteller des primären Antikörpers kein Fixiermittel vorschlägt, eignet sich für verschiedene Zelllinien und Antigene ein Start mit 4 % Formaldehyd für 10 Minuten bei Raumtemperatur.
Permeabilisierung
Durch die Permeabilisierung werden intrazelluläre Strukturen für Antikörper zugänglich, die andernfalls die Lipidmembranen der Zelle nicht passieren können. Je nach Art der Fixierung ist ein separater Permeabilisierungsschritt erforderlich. Bei der Fixierung mit organischen Lösungsmitteln sind die Zellmembranen bereits durchlässig und Sie können direkt mit der Blockierung fortfahren. Mit chemischen Vernetzern fixierte Zellen erfordern eine zusätzliche Behandlung mit einem Detergens zur Permeabilisierung. Dabei kommen klassische Detergenzien wie Triton X-100 oder NP-40 zum Einsatz, aber auch Saponin, Tween 20 oder Digitonin können verwendet werden. Auch hier werden unterschiedliche Ergebnisse erzielt, die von der verwendeten Substanz, ihrer Konzentration und der Inkubationszeit abhängen, so dass Sie zu Beginn verschiedene Parameter ausprobieren sollten. Ein typischer Start ist eine Permeabilisierung mit 0,1 % Triton X-100 in PBS für 15-20 Minuten bei Raumtemperatur.
Zur Analyse von Lipid-assoziierten Proteinen oder Membranproteinen mittels Immunoflurosenzen sollte der Permeabilisierungsschritt (= Lipidentfernung) sorgfältig durchgeführt werden. Eine gute Wahl für diesen Zweck ist Saponin, das selektiv Cholesterin aus der Plasmamembran entfernt und intrazelluläre Membranen weitgehend intakt lässt. Wird die Permeabilisierung vor der Antikörperfärbung weggelassen ( nur durch Fixierung mit chemischen Quervernetzern möglich), können extrazelluläre, an die Plasmamembran gebundene Antigene spezifisch markiert werden, wodurch sie vom intrazellulären Antigenpool unterschieden werden können. Nukleinsäurefarbstoffe wie DAPI oder Hoechst (siehe Nukleusfärbung und Probenmontage) sind membransensibel und erfordern keine Permeabilisierung.
Blockieren
Die Blockierung ist ein wesentlicher Schritt, um die unspezifische Bindung des primären Antikörpers in der Zelle zu minimieren. Um dies zu erreichen, können Proteine aus Rinderserumalbumin (BSA), Milchpulver oder Serum verwendet werden. Es ist wichtig, dass diese blockierenden Proteine nicht von der Spezies stammen, in denen der primäre Antikörper gebildet wurde, da sonst die Spezifität des sekundären Antikörpers gegenüber dem primären Antikörper verloren geht. Wenn Sie einen sekundären Antikörper verwenden, z. B. einen, der in der Ziege gegen einen murinen ersten Antikörper hergestellt wurde, ist normales Ziegenserum ein ideales Blockierungsreagenz. Blockierungslösungen werden normalerweise in Konzentrationen von 1 % (Milchpulver, BSA) bis 5 % (normales Serum) verwendet und in Waschpuffer verdünnt. Die Inkubation erfolgt bei Raumtemperatur für 30-60 Minuten.
Immunreaktion
Nachdem die Proben durch Fixierung, Permeabilisierung und Blockierung vorbereitet wurden, findet die eigentliche Immunreaktion statt. Die Proben werden nun mit dem spezifischen Primärantikörper inkubiert, um die gewünschten Zielstrukturen zu markieren. Es können mehrere primäre Antikörper gleichzeitig auf die Probe aufgebracht werden. Wie bereits erwähnt, müssen bei der indirekten Multicolor-Immunfluoreszenz (IF) mehrere Primärantikörper von verschiedenen Spezies stammen. Die Auflösung der Antikörper erfolgt in der von Ihnen gewählten Blockierungslösung, zunächst entsprechend den Angaben des Herstellers. Wenn Sie mit Ihrer Färbung unzufrieden sind oder der Hersteller keine Angaben zu einer geeigneten Verdünnung macht, sollten Sie Konzentrationen zwischen 1:50 und 1:1.000 ausprobieren. Je nach der Affinität des Antikörpers kann die Inkubationszeit variieren. Die Standard-Inkubationszeit beträgt 1-2 Stunden bei Raumtemperatur; eine Inkubation über Nacht bei 4 °C ist ebenfalls möglich.
Bei der direkten Immunfluoreszenz können Sie direkt mit der Probenaufbereitung fortfahren, da der primäre Antikörper bereits sein eigenes Fluorochrom mitbringt. Bei der indirekten Immunfluoreszenz markieren die sekundären Antikörper nun die primären Antikörper fluoreszierend. Ein entscheidender Punkt ist hier das umfangreiche Waschen nach der Inkubation mit dem primären Antikörper, damit die unspezifische Bindung des sekundären Antikörpers reduziert wird. Der Sekundärantikörper kann auch in Blockinglösung oder Waschpuffer verdünnt werden. Wenn vom Hersteller nicht anders angegeben, können Sie mit einer Verdünnung von 1:200 und einer Inkubation von 1 Stunde bei Raumtemperatur beginnen. Die Inkubationsschritte müssen im Dunkeln durchgeführt werden, um ein Ausbleichen des Fluorochroms zu verhindern.
Zellkernfärbung und Probenmontage
Auf die Immunreaktion folgt häufig eine Kernfärbung mit DNA-Farbstoffen. Dies ermöglicht zum einen eine bessere Orientierung innerhalb der Zelle oder der Gewebeschnitte bei der Mikroskopie und zeigt zum anderen den Zellstatus (z.B. Mitose) an, wenn dies für den Forscher von Interesse ist. Dazu werden Farbstoffe wie Hoechst oder DAPI verwendet, die auch ohne Permeabilisierung in den Zellkern gelangen und sich in die DNA einlagern. Aus diesem Grund sollte man sehr vorsichtig sein und den direkten Hautkontakt mit diesen Farbstoffen vermeiden! Eine einfache Kernfärbung erfolgt für 10 Minuten bei Raumtemperatur mit in PBS verdünntem Hoechst oder DAPI.
Nach Abschluss des Immunfluoreszenzverfahrens (IF) müssen die Proben für die Mikroskopie eingebettet werden. Zu diesem Zweck wird ein Einbettungsmedium (z. B. Mowiol oder Prolong Gold) verwendet, das die Probe auf einem Objektträger fixiert und gleichzeitig ihre Austrocknung verhindert. Außerdem erhöhen Einbettmittel den Brechungsindex, was für die Mikroskopie mit Hilfe von Objektiven mit Ölimmersion förderlich ist. Mehrere Hersteller bieten Einbettmittel mit Zusätzen wie DABCO an, einem Antiverblendungsmittel, das die Probe vor dem Ausbleichen durch Licht schützt. Je nach den verwendeten Fluorochromen sind einige Anti-Fading-Mittel wirksamer als andere. Darüber hinaus sind auch Einbettmittel mit zugesetzten DNA-Farbstoffen erhältlich, so dass die Kerne während des Einbettens angefärbt werden und eine separate Kernfärbung überflüssig ist. Wird ein aushärtendes Einbettungsmittel verwendet, was häufig der Fall ist, lässt man die Probe über Nacht aushärten, so dass die mikroskopische Untersuchung am nächsten Tag möglich ist. Die so hergestellten Dauerpräparate können praktisch unbegrenzt im Dunkeln bei Raumtemperatur oder 4 °C gelagert werden, wobei zu beachten ist, dass die Fluoreszenzintensität der Fluorochrome mit der Zeit abnimmt.
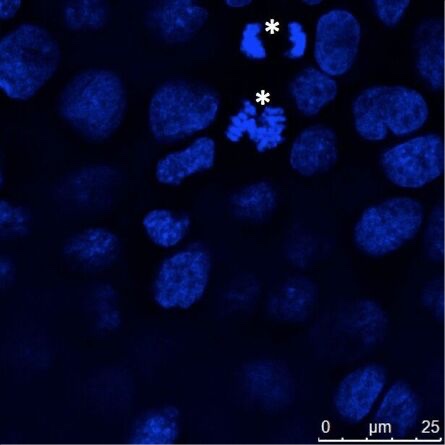
Abb. 4: Adhärent wachsende Epithelzellen (MDCK) wurden auf Deckgläsern kultiviert, fixiert und die Zellkerne mit Hoechst 33342 gefärbt. Die meisten Zellen weisen eine Interphasen-DNA-Färbung auf, aber einige Zellen zeigen kondensierte Chromosomen, die in der Mitose getrennt werden (Sternchen). Die Mikroskopie wurde mit einem Leica TCS SP2 durchgeführt.
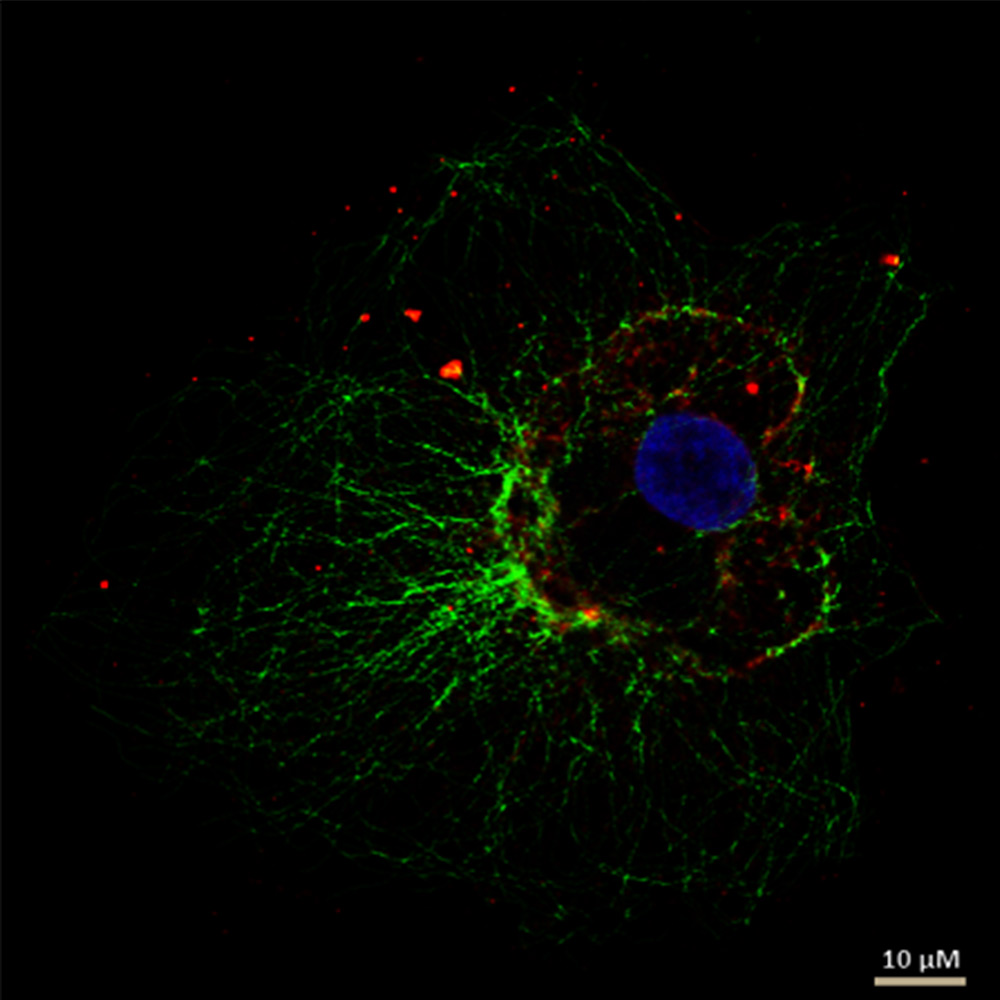
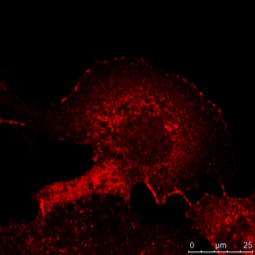
Abb. 5: Dieses Bild zeigt eine indirekte Immunfluoreszenz-Färbung des zellulären Mikrotubuli-Netzwerks in einer Fibroblastenzelle (COS-7). Der Zellkern wurde mit Hoechst 33342 angefärbt und die Mikroskopie wurde mit einem Leica TCS SP2 durchgeführt.
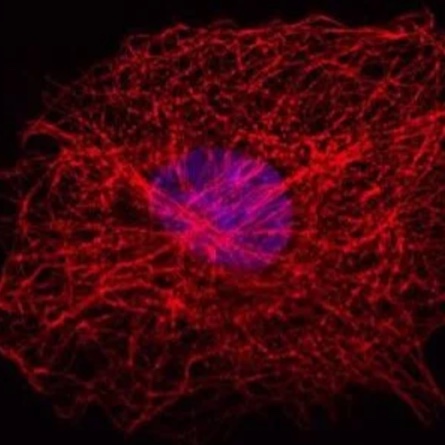
Abb. 4: Adhärent wachsende Epithelzellen (MDCK) wurden auf Deckgläsern kultiviert, fixiert und die Zellkerne mit Hoechst 33342 gefärbt. Die meisten Zellen weisen eine Interphasen-DNA-Färbung auf, aber einige Zellen zeigen kondensierte Chromosomen, die in der Mitose getrennt werden (Sternchen). Die Mikroskopie wurde mit einem Leica TCS SP2 durchgeführt.
Abb. 5: Dieses Bild zeigt eine indirekte Immunfluoreszenz-Färbung des zellulären Mikrotubuli-Netzwerks in einer Fibroblastenzelle (COS-7). Der Zellkern wurde mit Hoechst 33342 angefärbt und die Mikroskopie wurde mit einem Leica TCS SP2 durchgeführt.
Steuerung
Entscheidend für die Immunfluoreszenz sind entsprechende Kontrollen für die korrekte Interpretation Ihrer Mikroskopieaufnahmen. Fehlende oder falsche Kontrollen führen in der Regel zu falsch positiven Aussagen und falschen Daten. Zu Beginn sollten Sie eine Probe analysieren, die nur fixiert, permeabilisiert und blockiert wurde, um eine Referenz der Autofluoreszenz der zellulären Kompartimente zu erhalten. Manchmal erscheinen Strukturen stark fluoreszierend, obwohl keine Färbung durch Immunfluoreszenz vorliegt.
Als weitere Kontrolle und zur Einstellung der Mikroskopieparameter für die indirekte Immunfluoreszenz (IF) wird eine Probe verwendet, die ebenfalls wie oben beschrieben behandelt, aber zusätzlich mit dem/den sekundären Antikörper(n) inkubiert wurde. Erstens würde dies eine starke unspezifische Bindung des sekundären Antikörpers an die Probe aufzeigen. Zweitens werden die Mikroskopieparameter so eingestellt, dass bei der Bildaufnahme dieser Kontrolle kein Signal aufgezeichnet wird. Diese Einstellung wird als Schwellenwert für nachfolgende Aufnahmen verwendet, um falsch-positive Signale durch den sekundären Antikörper auszuschließen. Bei der direkten Immunfluoreszenz (IF) dient die Autofluoreszenzkontrolle zur Einstellung der Schwelle. Anschließend analysieren Sie die Proben, die mit dem spezifischen primären Antikörper und im Falle der indirekten Immunfluoreszenz (IF) auch mit dem sekundären Antikörper inkubiert wurden. Hier sollte nun ein Fluoreszenzsignal nachweisbar sein, das oberhalb der Autofluoreszenz und der Negativkontrolle des sekundären Antikörpers liegt.
Um sicher zu sein, dass der erste Antikörper ausschließlich die gewünschten Strukturen markiert, bieten einige Hersteller ein blockierendes Peptid für den ersten Antikörper an, mit dem das spezifische Antigen maskiert wird, so dass der Antikörper nicht an sein Epitop binden kann. Dies ist zwar teurer, aber auch der beste Weg, um die Spezifität des Antikörpers zu bestimmen und somit zuverlässige Ergebnisse zu erhalten. Bei der Durchführung eines Multicolor-Immunofluoreszenzexperiments müssen Sie auch auf das Übersprechen zwischen den ausgewählten Fluorochromen achten. Führen Sie zum ersten Mal eine Multicolor-Immunofluoreszenz durch, ist es ratsam, die Zielstrukturen zusätzlich in separaten Präparaten zu färben und diese Bilder mit dem Multicolor-Bild zu vergleichen. Letztendlich sollten Sie Ihre gewonnenen Daten genau betrachten und mit Ihren Erwartungen und vorhandenen Daten zum gleichen Primärantikörper vergleichen.
Einschränkungen
Wie bereits beschrieben, hat die Immunfluoreszenz-IF viele Vorteile, bringt aber auch einige Nachteile mit sich. Ein wichtiger Punkt ist die Fixierung der Probe: Fixierung bedeutet Abtöten, wodurch die Darstellung lebender Zellen nicht mehr möglich ist. Die Auswertung von dynamischen Prozessen wird dadurch erschwert, indem für jeden Zeitpunkt Zellen fixiert und angefärbt werden müssen. Schnelle dynamische Prozesse sind also mit Immunfluoreszenz nicht zu beobachten. Dies ist eindeutig ein Vorteil der Expression von Fusionsproteinen mit fluoreszierenden Markierungen wie GFP, die sich für die Bildgebung in lebenden Zellen eignen. Wie bereits erwähnt, ändert das Immunfluoreszenz-Verfahren (Fixierung/Permeabilisierung) die zelluläre Architektur, so dass Artefakte als falsch positive Signale interpretiert werden können. Es ist daher unerlässlich, für jede Immunfluoreszenz-Färbung geeignete Kontrollen vorzubereiten, was zeitaufwändig sein kann. Ein weiterer Nachteil ist unvermeidlich: das Photobleaching der Fluorochrome. Fluoreszierende Proteine wie GFP sind davon ebenfalls betroffen, aber bei ordnungsgemäßer Lagerung ist GFP in Dauerpräparaten auch nach Monaten noch nachweisbar. Im Gegensatz dazu verlieren die IF-Fluorochrome ihre Intensität schneller, was sich durch ein schnelles Ausbleichen der Probe während der Mikroskopie bemerkbar macht. Auch Trägermaterial mit Anti-Fading-Mitteln kann hier nur vorübergehend helfen.
Standard-Immunfluoreszenzprotokoll
Dauer eines Immunofluoreszenzverfahrens: ca. 5 Stunden.
Dies ist ein Standardprotokoll für die indirekte Immunfluoreszenz von kultivierten Zellen auf Deckgläsern mit Fixierung durch chemische Vernetzer
- Eine befeuchtete Kammer ist ideal für das Immunfluoreszenzverfahren und kann leicht selbst hergestellt werden (siehe Diashow "Vorbereitung einer befeuchteten Kammer"). Sie verhindert das Austrocknen des Präparats und ermöglicht die Inkubation im Dunkeln, was beim Umgang mit Fluorochromen wichtig und bei bereits vorhandenen fluoreszierenden Proteinen notwendig ist.
- Die Volumina werden so gewählt, dass die Deckgläser vollständig befeuchtet werden. Achten Sie darauf, dass die Probe nie ganz trocken wird.
- Alle Inkubationsschritte finden bei Raumtemperatur statt
- …
Rezepte
Waschpuffer
- 1 × PBS (Phosphatpuffersalzlösung)
- 137 mM: NaCl
- 2.7 mM: KCl
- 10 mM: Na2HPO4
- 1.8 mM: KH2PO4
- Stellen Sie den pH-Wert mit HCl auf 7,2-7,4 ein.
- Für PBS++ eine Endkonzentration von 1 mM CaCl2 und MgCl2 hinzufügen
- Für PBS-T eine Endkonzentration von 0,05 % Tween 20 hinzufügen
Fixierpuffer
- Formaldehyd:
- 4 % PFA (Paraformaldehyd) in warmem (50–70 °C) dH2O bei pH 8 (mit NaOH einstellen) auflösen.
- 10 × PBS bis zu einer Endkonzentration von 1 × PBS hinzufügen (z. B. 100 mL 10 × PBS in 900 mL 4 % PFA/dH2O).
- pH-Wert mit HCl auf 7,2-7,4 einstellen.
- Methanol (vorgekühlt auf -20 °C):
- 100 % Methanol (-20 °C)
- Methanol/Aceton (vorgekühlt auf -20 °C):
- 50 % Methanol (–20 °C) and 50 % Acetone (–20 °C)
Permeabilisierungspuffer
- TX-100 (Triton X-100):
- PBS mit einer Endkonzentration von 0,1% TX-100
- Saponin:
- PBS mit einer Endkonzentration von 0,1 % Saponin
- Andere Detergenzien können in der gleichen Konzentration in PBS verwendet werden.
Blockierpuffer
- BSA (Rinderserumalbumin):
- PBS mit einer Endkonzentration von 1 % BSA
- Milchpulver:
- PBS mit einer Endkonzentration von 1 % Milchpulver
- Normales Serum:
- PBS mit einer Endkonzentration von 5 % normalem Serum
Nukleäre Farbstoffe
- Hoechst oder DAPI:
- Bereiten Sie eine Lösung von Hoechst 33342 oder DAPI mit einer endgültigen Arbeitskonzentration von 1 µg/ml in PBS vor.
 and phalloidin (magenta), imaged using Viventis SCAPE; scale bar 50μm. Courtesy of Marina Cuenca and Heleen Jungen (Dayton lab), EMBL Barcelona.")



