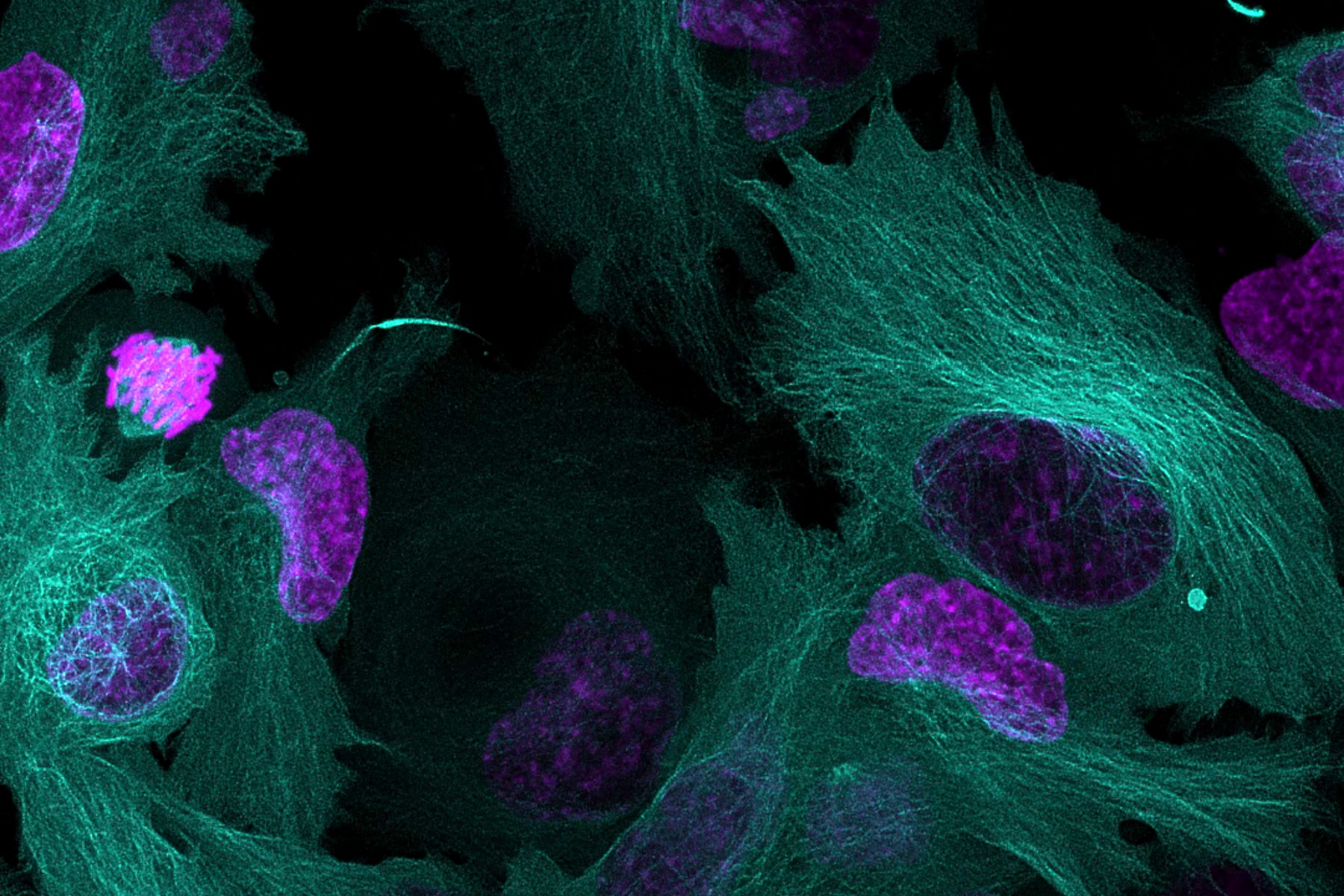
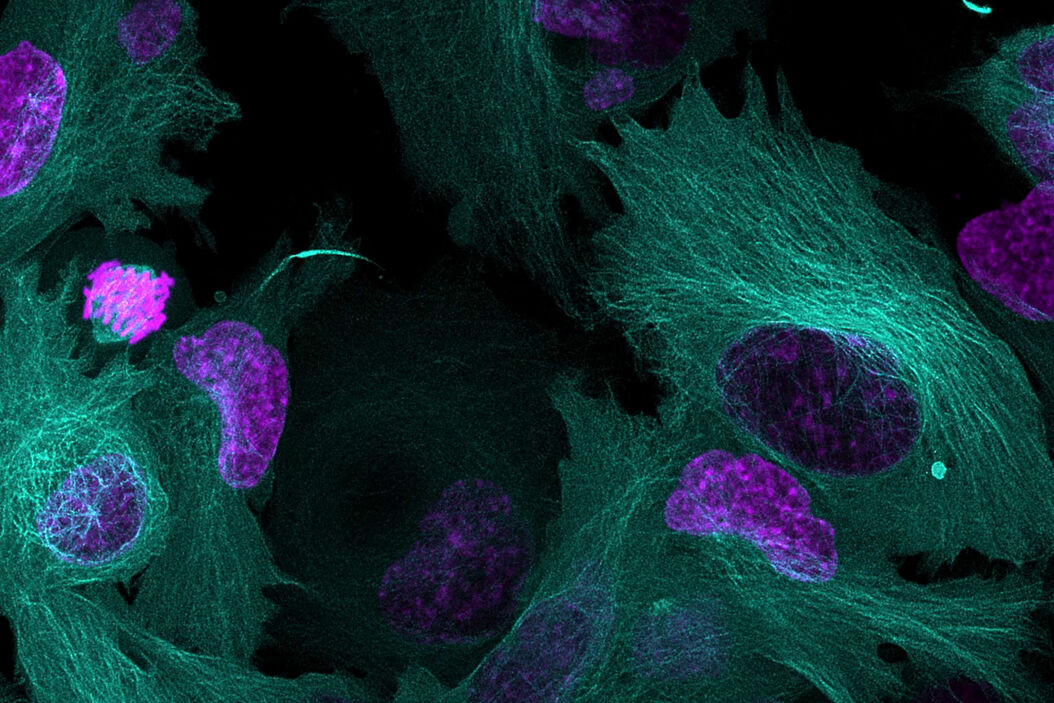
Why is it important to use more than one fluorescent marker?
Multiplexing in live-cell microscopy allows us to observe multiple cell structures and processes in synchrony, providing more physiologically relevant results and thus adding context to measurements. It allows the study of cell and protein interactions, dynamic changes, and how these changes interact and influence each other. However, while experiments using a single fluorophore are common, imaging live cells using two or more fluorescent probes can be complex and requires the consideration of several important points to be successful. This article will cover some key aspects of setting up a successful multicolor live-cell experiment.
Download the eBook to discover some key aspects of setting up a successful multicolor live cell experiment as well as avoiding some of the major pitfalls. Plus, find out how the Mica imaging Microhub is highly adapted for live cell imaging experiments.
See the difference that Mica could make to your multiplex live cell imaging experiment!
What you will learn
Key learnings
- Selecting the right fluorophore combinations
- Avoiding the misleading effects of fluorescence crosstalk
- Maintaining near physiological conditions
- Considerations for image acquisition

Selecting the right fluorophores
When setting up a multicolor live-cell experiment, first it is crucial to choose the correct fluorophores. They must be compatible with live specimens and photostable to allow long-term imaging. Fluorescent protein tags, live-cell dyes, dynamic markers like Ca2+ indicators, and other methods used for labeling the target of interest are the right tools for this purpose. Nevertheless, the choice of fluorescent dyes to study processes in living cells in a multiplexed manner is still quite limited due to broad emission spectra. With each additional label used, it becomes increasingly difficult to find excitation/detection schemes with high specificity and high signal yield. Complex labeling strategies are needed to minimize spectral overlap when two or more fluorophore signals are imaged together.
Imaging under physiological conditions
Whether you are using cell cultures, organoids, spheroids, or small model organisms, you must ensure that the sample is kept alive by using near physiological conditions. In time-lapse experiments, certain conditions must be kept optimal to ensure not only that the cells survive, but also that they are not stressed and retain their true physiology. Usually, an incubator (37 °C, CO2, humidity, and sometimes O2 with high stability over time) is used to ensure constant near physiological conditions. It also pays to choose an imaging compatible medium that has low autofluorescence and does not interfere with the signals.
Considerations for image acquisition
Widefield microscopy is usually the first choice for imaging living cells because of its faster and gentler imaging performance. State-of-the-art widefield microscopy uses LED light sources and optical filter sets. Although there is a wide range of filters available that allow for the use of a variety of fluorophore combinations, several things need to be considered to get the correct images from samples labeled with several fluorophores.
Most fluorophores used for live-cell imaging have broad emission spectra. This fact often means that you get crosstalk between detection channels, e.g., you see some emission from the green channel in the red detection channel. As fluorescent spectral overlap is common when using multiple labels in live-cell imaging experiments, to ensure you see the details you are trying to identify, you can use narrow bandpass filters. However, this approach results in reduced sensitivity and compromise the results, as the fluorophore signal levels in live cells are often low, especially for specimens with sparse target abundance or those expressed at endogenous levels. Counteracting the reduced sensitivity by using more excitation light is often not possible as it is increasingly harmful (i.e. increased phototoxicity) for the sample.
For live cell experiments, high speed acquisition is often critical, in particular for the study of fast dynamic processes such as vesicle observation. Using optical filters results in speed limitations due to the necessity for sequential imaging when changing filter sets for each color. Gathering images sequentially requires more time than simultaneous image gathering and, as a result, rapid specimen motions can be missed during acquisition, as each color has a longer time interval from one image to the next. On top when the direct comparison between two or more colors is of essence, as in the above-mentioned vesicle observation experiment: the signals may have moved even between the individual acquisition of the fluorophores, complicating the interpretation of the data.
Alternatives for improving live-cell imaging
Because the amount of information obtained by studying the interaction between multiple probes in live cells leads to more physiologically relevant results, methods have been developed to unravel complex mixtures of emission signals.
To enable the use of multiple labels and, therefore, increase the insights you get from one sample, you can use linear unmixing of spectral image information, a method that analyses the contribution of each fluorophore to the total signal. To perform linear unmixing, the minimum difference between the unknown and reference spectra for all possible mixtures of the reference spectra must be calculated (1). This approach is commonly used to image multiple colors even when they overlap. However, for widefield microscopy, you might still miss critical information, as speed limitations result from the necessity to perform sequential image acquisition for each fluorophore in the specimen.
What if there were another way?
There is a method that allows for the rapid unmixing of multiple colors without having to worry about spectral overlap during acquisition or the need to learn complex unmixing approaches.
Recent publications show a phasor-based approach to unmixing (2, 3). Spectral phasor analysis transforms the spectral composition of each pixel into a location in a 2D plot. This way, emissions from different dyes get separated on the fly and reliably using simple math. Additionally, it allows to add filtering purely based on the spectrum rather than on the intensity - retaining fine details in the image while reducing noise that linear unmixing would suffer from. Imaging this way makes it easier to set up multiplex live cell imaging experiments, as selecting the right combination of fluorescent probes is no longer a problem thanks to the virtual elimination of crosstalk or even unwanted contributions such as autofluorescence.
 and phalloidin (magenta), imaged using Viventis SCAPE; scale bar 50μm. Courtesy of Marina Cuenca and Heleen Jungen (Dayton lab), EMBL Barcelona.")