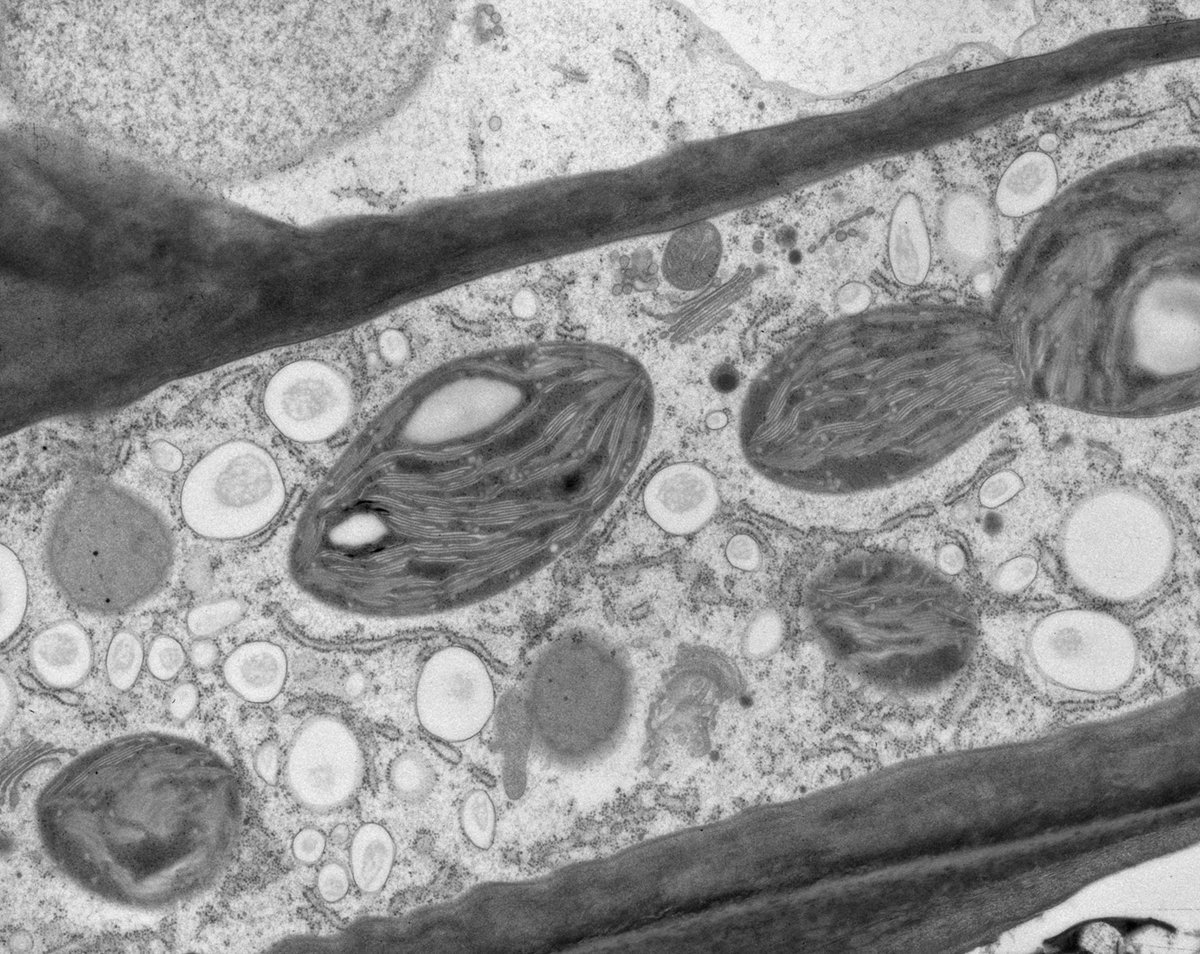
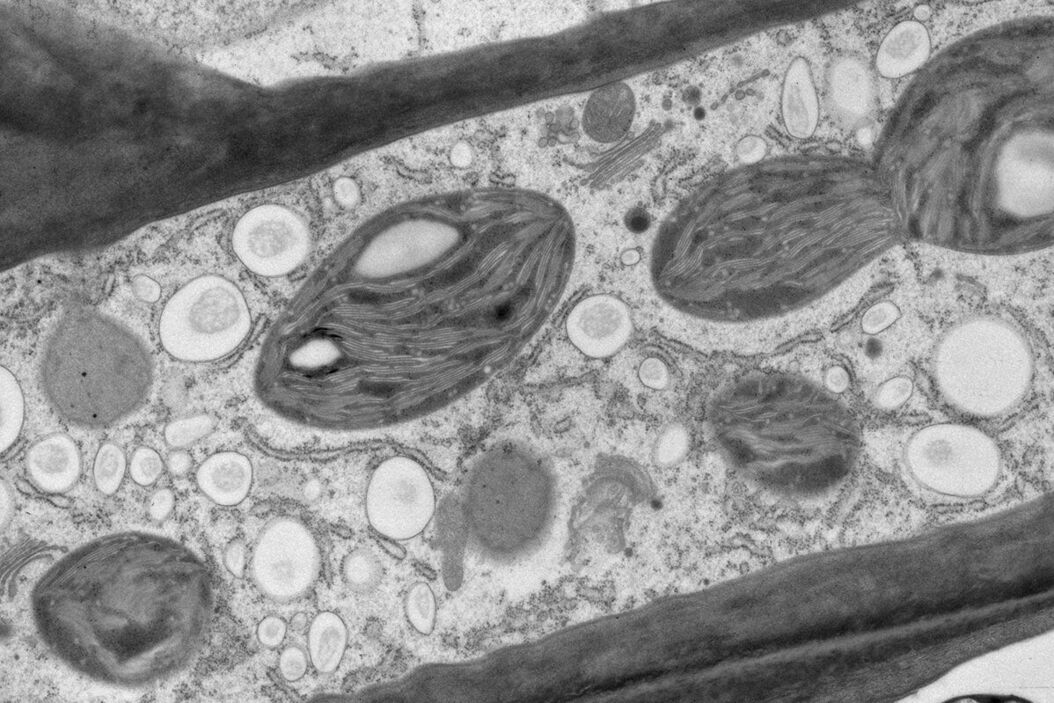
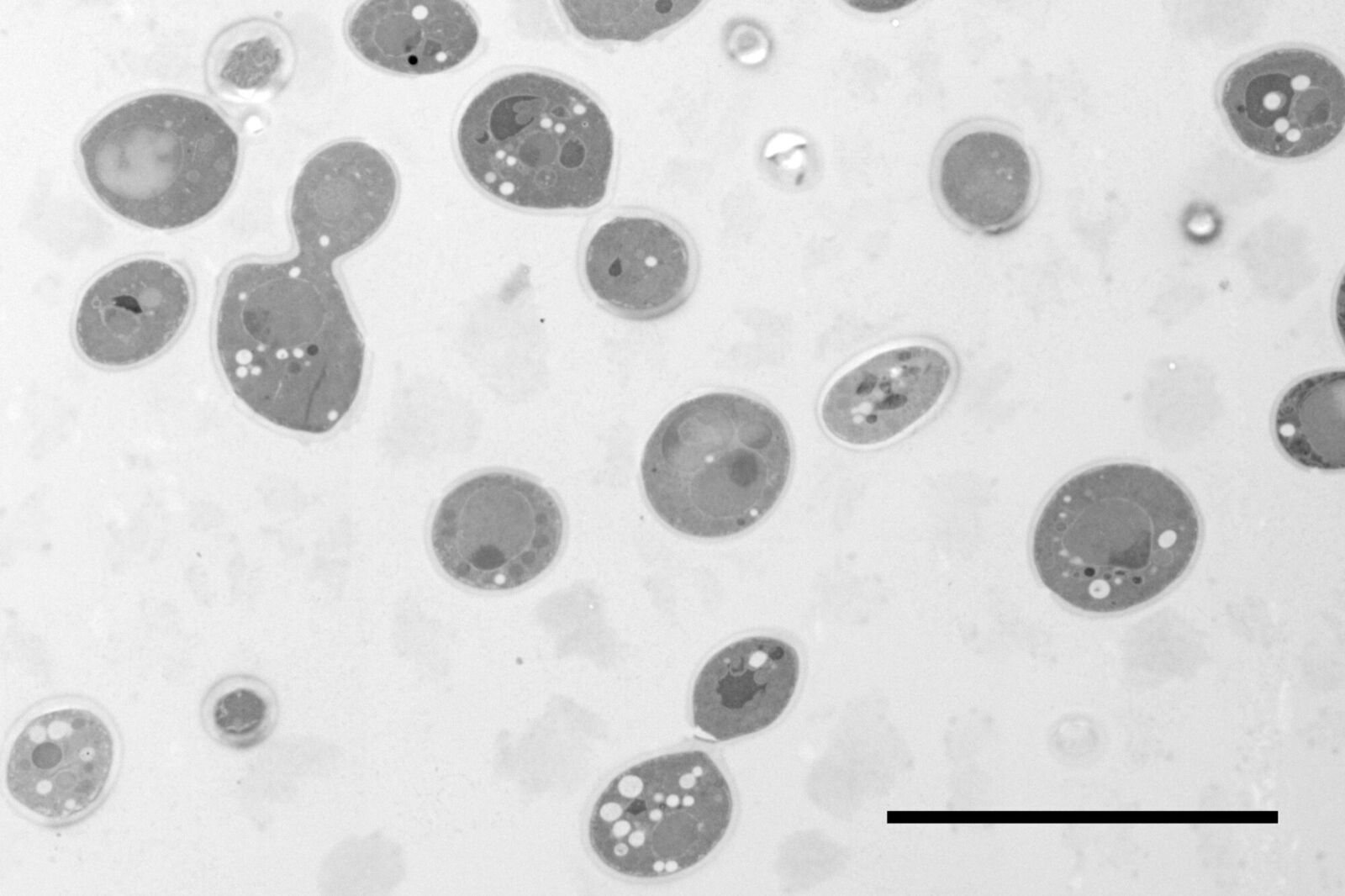
Fig. 1: Yeast (S. cerevisiae). Courtesy of van Donselaar EG, Humbel BM, Utrecht University, The Netherlands; Slot JW, University Medical Center, Utrecht, The Netherlands.
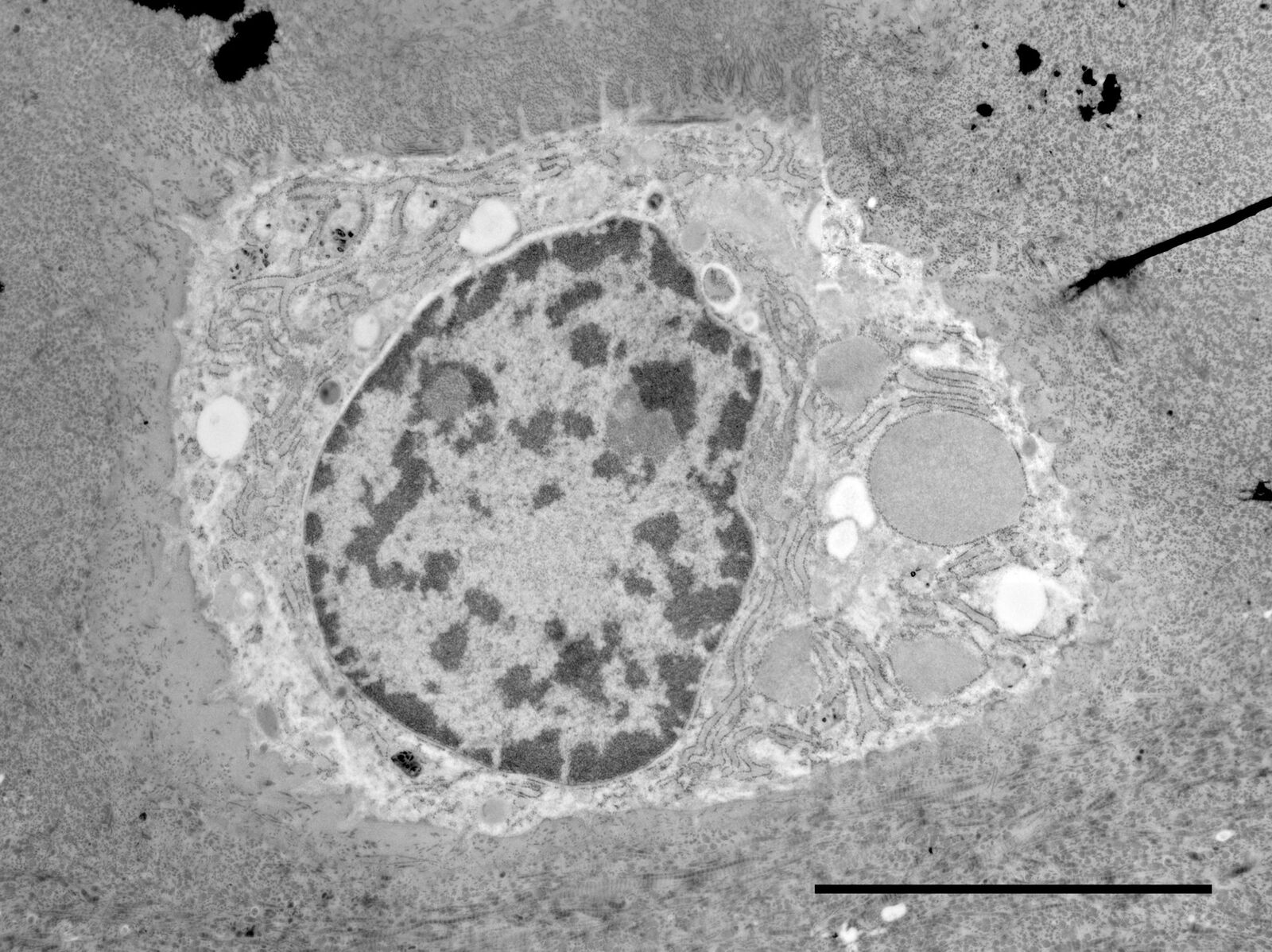
Electron microscopy of high pressure frozen and freeze substituted Arabidopsis thaliana root tips cells
Courtesy: de Rycke R
Protocol HPF – AFS for morphological analysis
Arabidopsis thaliana roots (mutant PIN1pro:PIN1-GFP;bex5-1) were excised, immersed in 20 % (w/v) BSA and frozen immediately in a high-pressure freezer (Leica EM PACT).
Freeze substitution was carried out using a Leica EM AFS2 in dry acetone containing 1 % ddH2O, 1 % OsO4 and 0.5 % glutaraldehyde over a 4-days period as follows:
- –90 °C for 24 hours,
- 2 °C per hour increase for 15 hours,
- –60 °C for 24 hours,
- 2 °C per hour increase for 15 hours, and
- –30 °C for 24 hours.
Samples were then washed 3 times in pure acetone between –30 °C and 0 °C and slowly warmed up to 4 °C, infiltrated stepwise over 3 days at 4 °C in Spurr’s resin and embedded in capsules. The polymerization was performed at 70 °C for 16 h.
Ultrathin sections were made using an ultramicrotome (Leica EM UC6) and post-stained in in a Leica EM AC20 for 40 min in uranyl acetate at 20 °C and for 10 min in lead citrate at 20 °C. Grids were viewed with a JEM 1010 transmission electron microscope (JEOL, Tokyo, Japan) operating at 80 kV using Image Plate Technology from Ditabis (Pforzheim, Germany).
For details see [5].
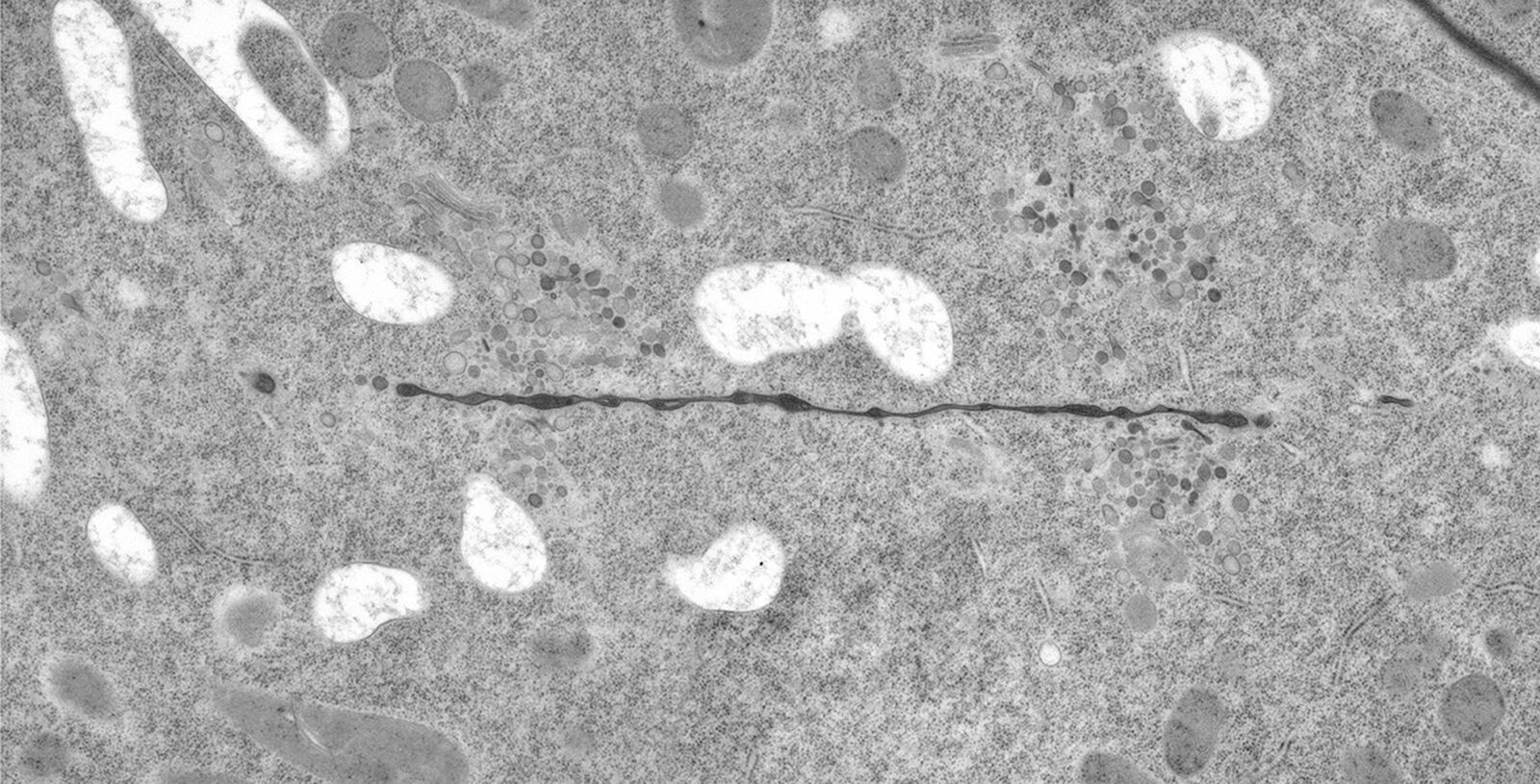
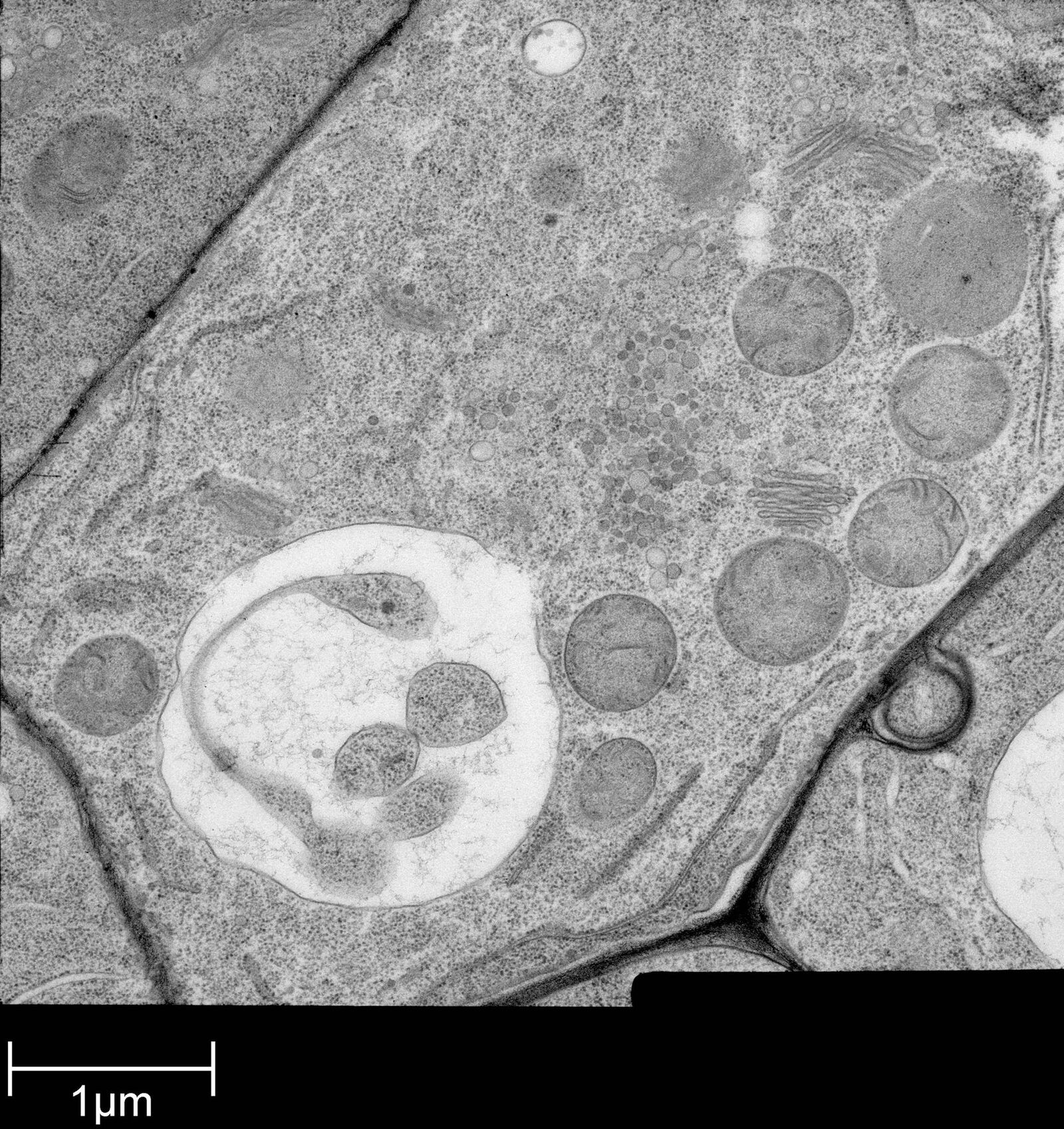
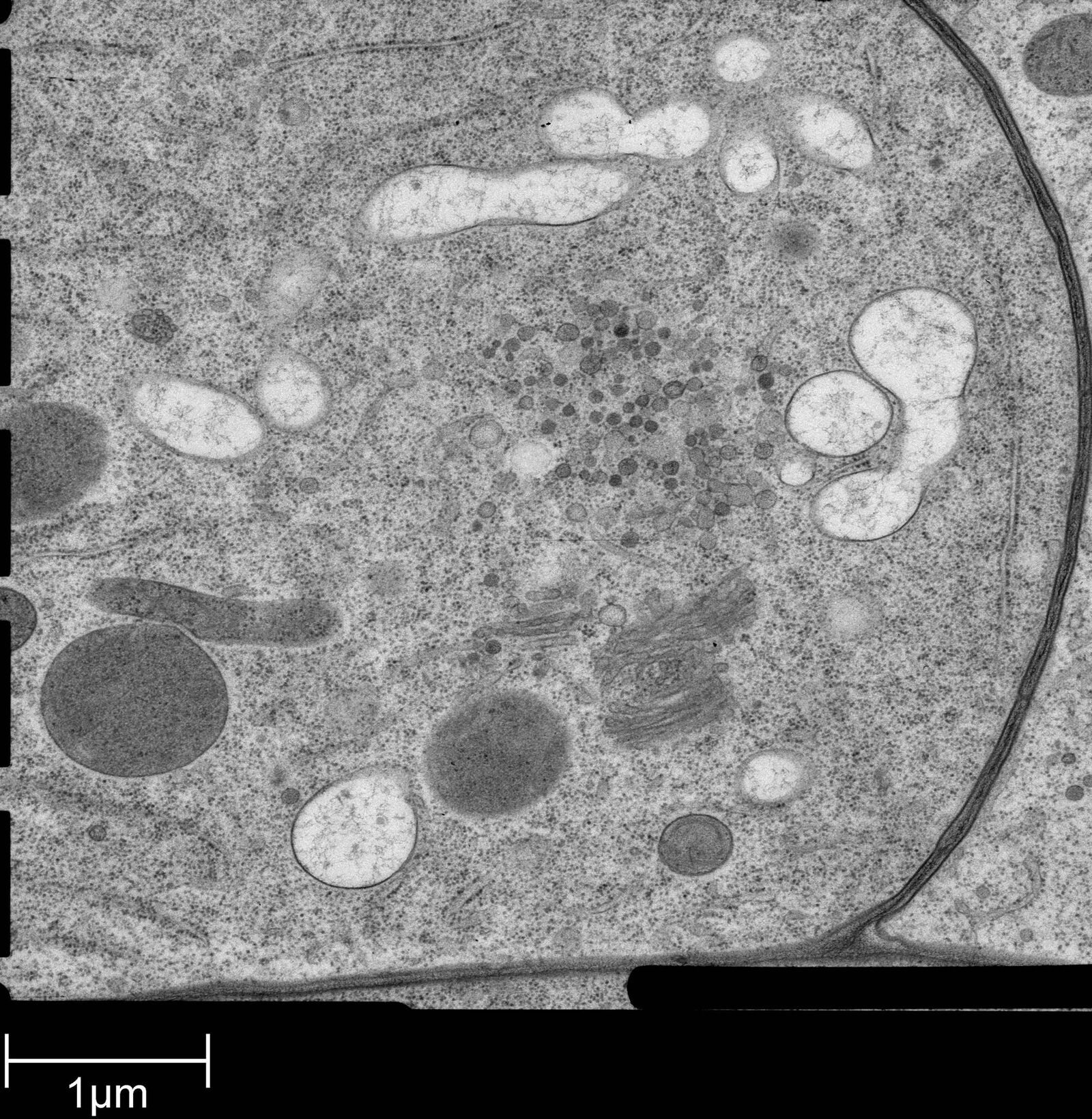
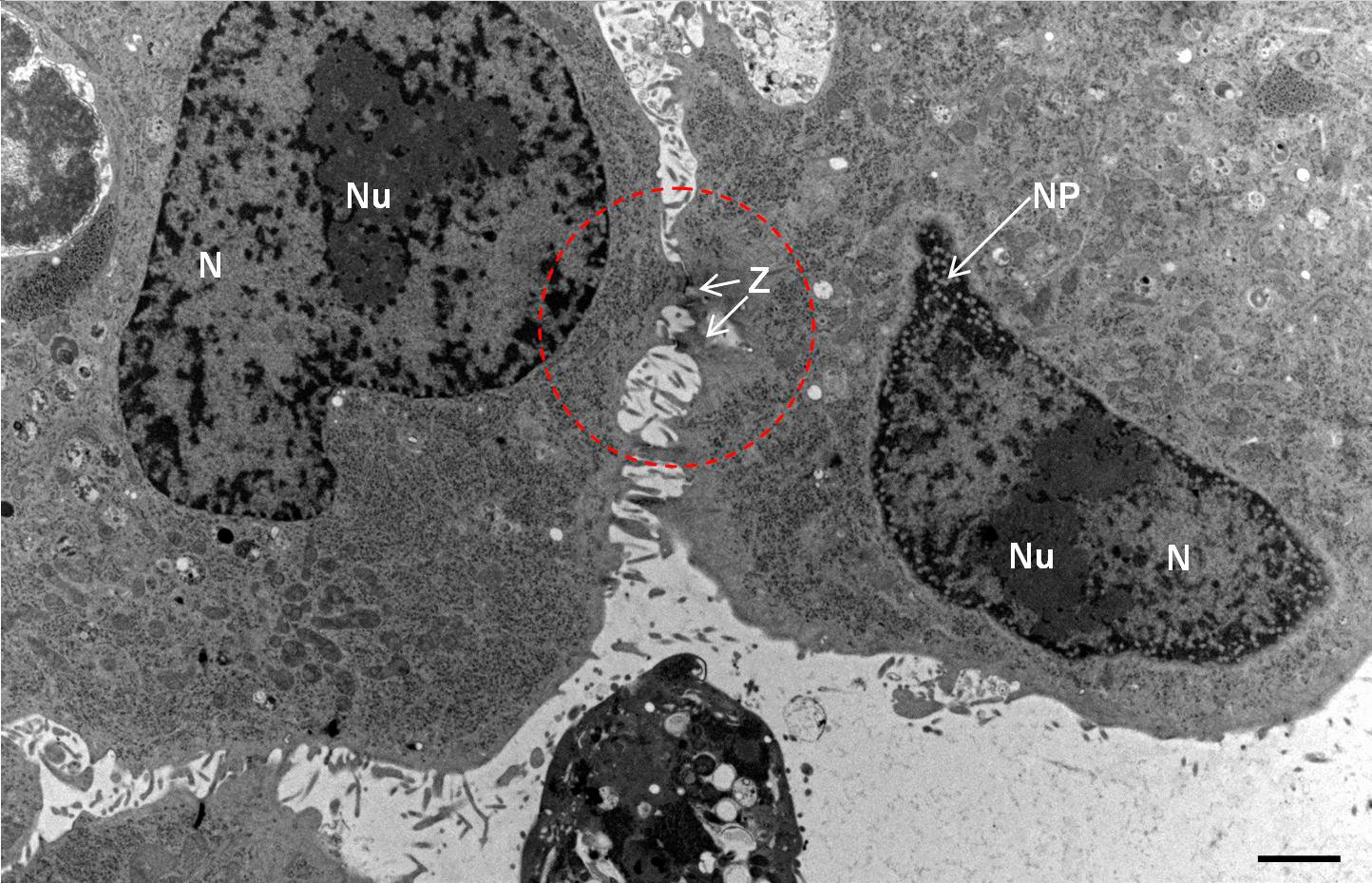
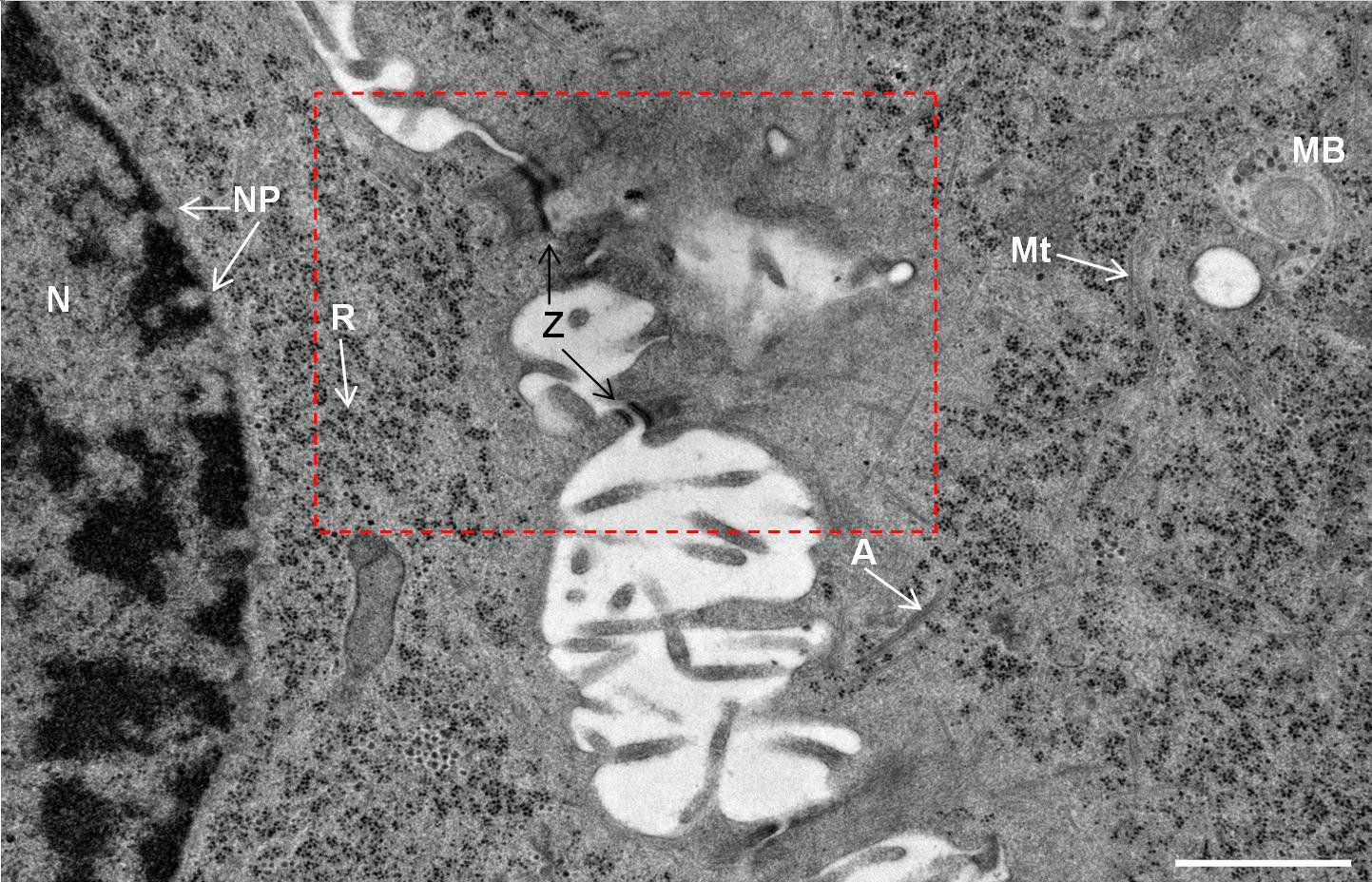
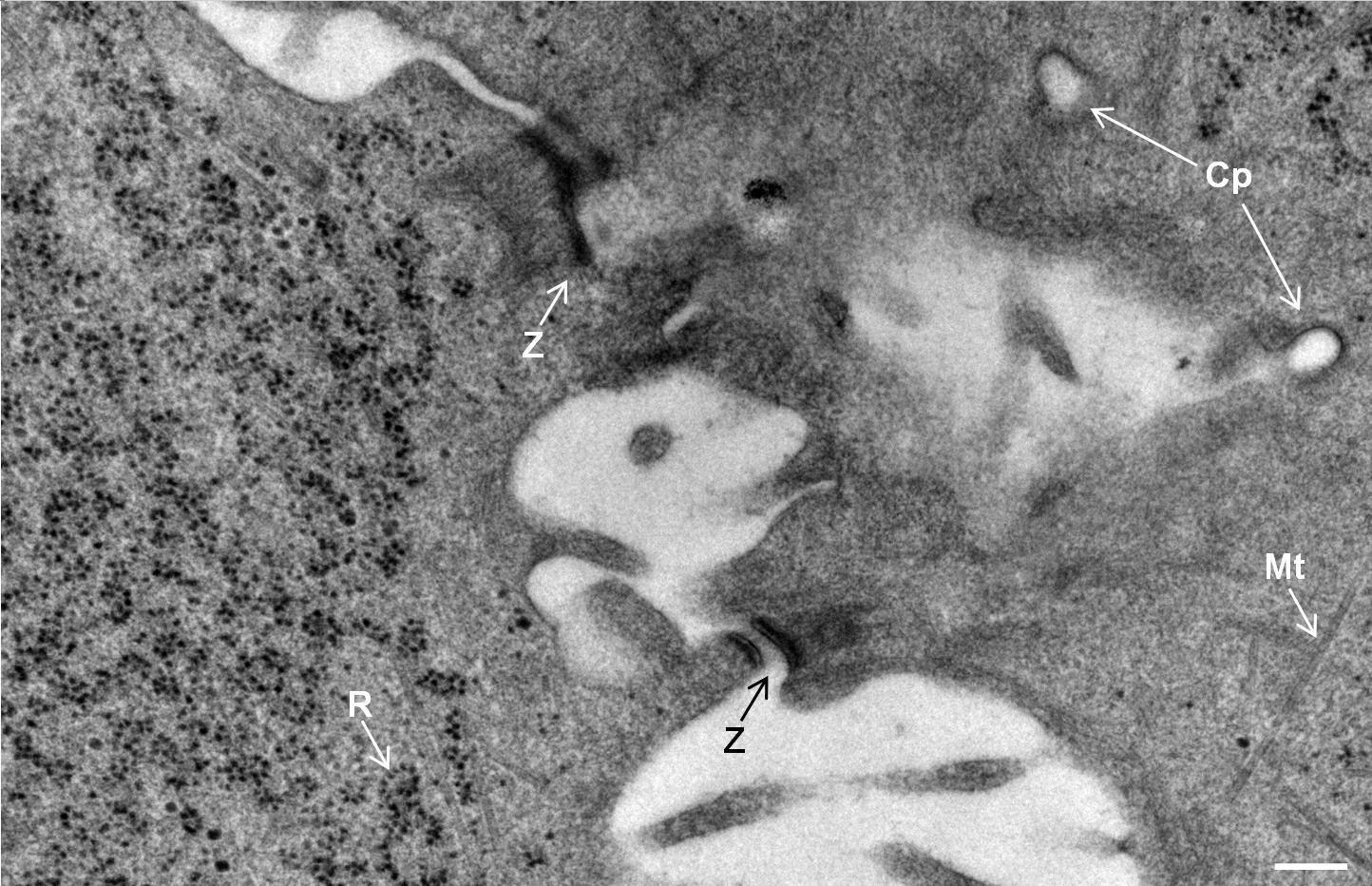
Fig. 2: Ultrastructure of Arabidopsis thaliana primary root cells in PIN1pro::PIN1=GFP;bex5-1 treated for 1 h with 50 uM BFA.
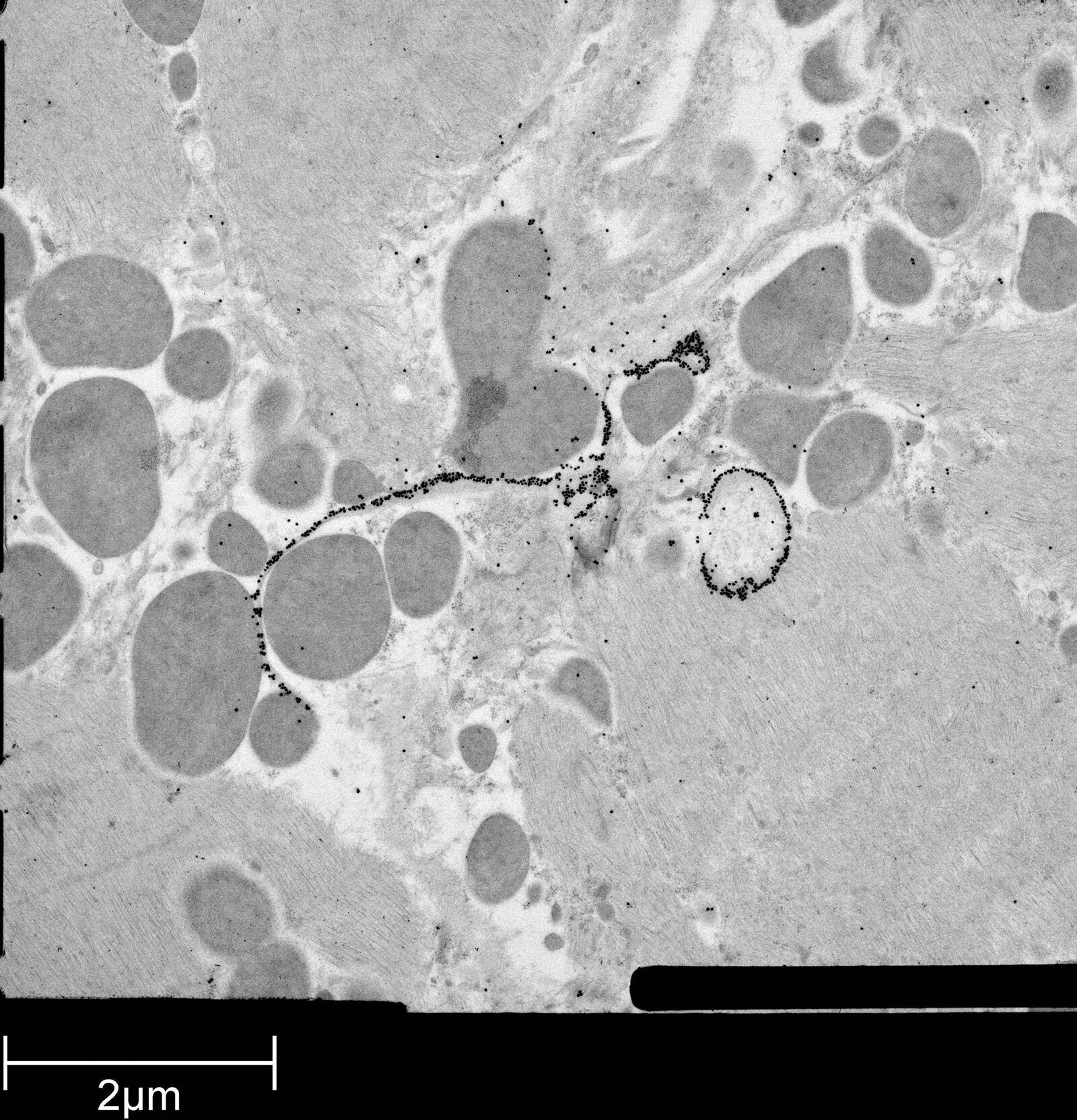
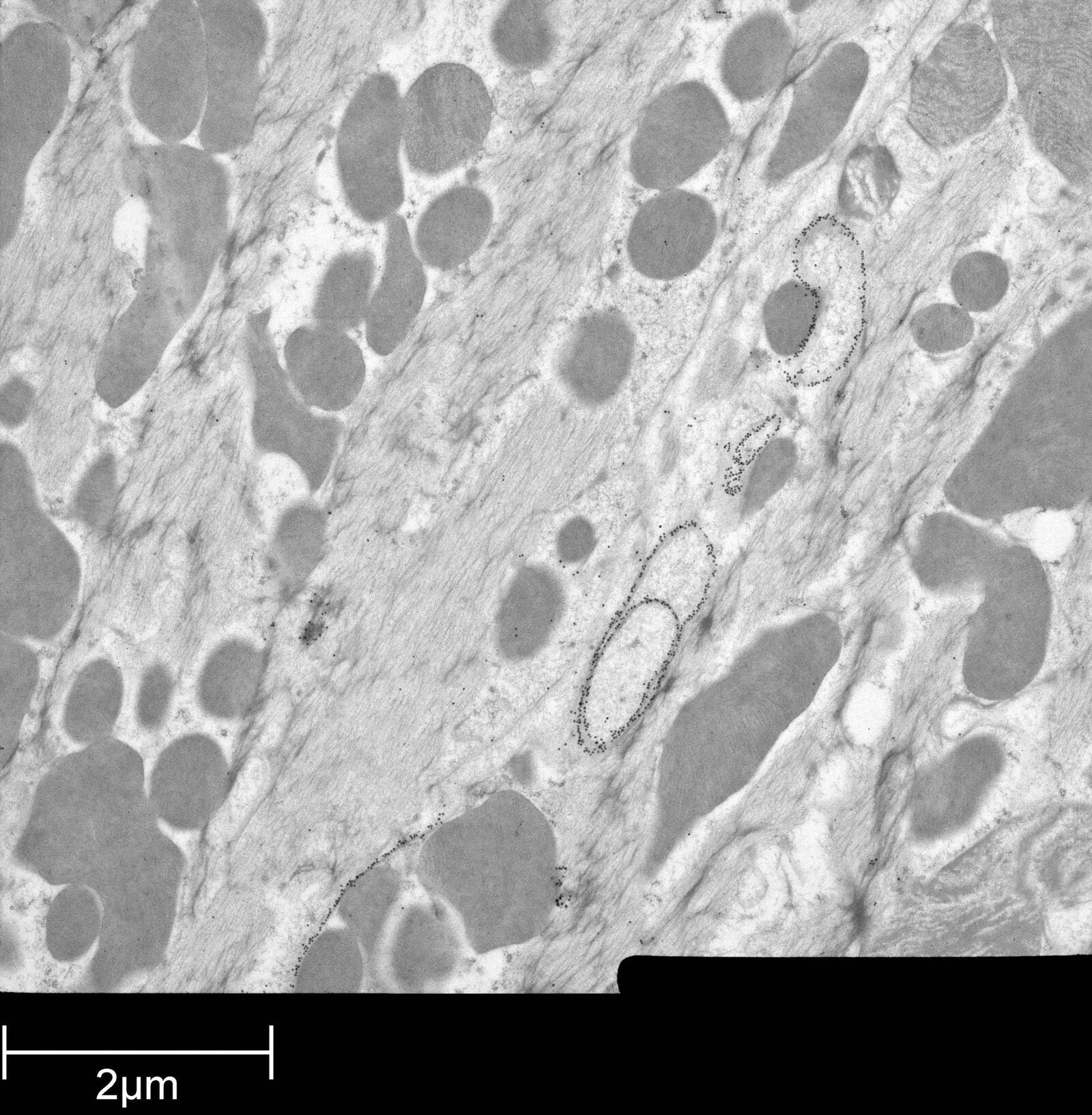
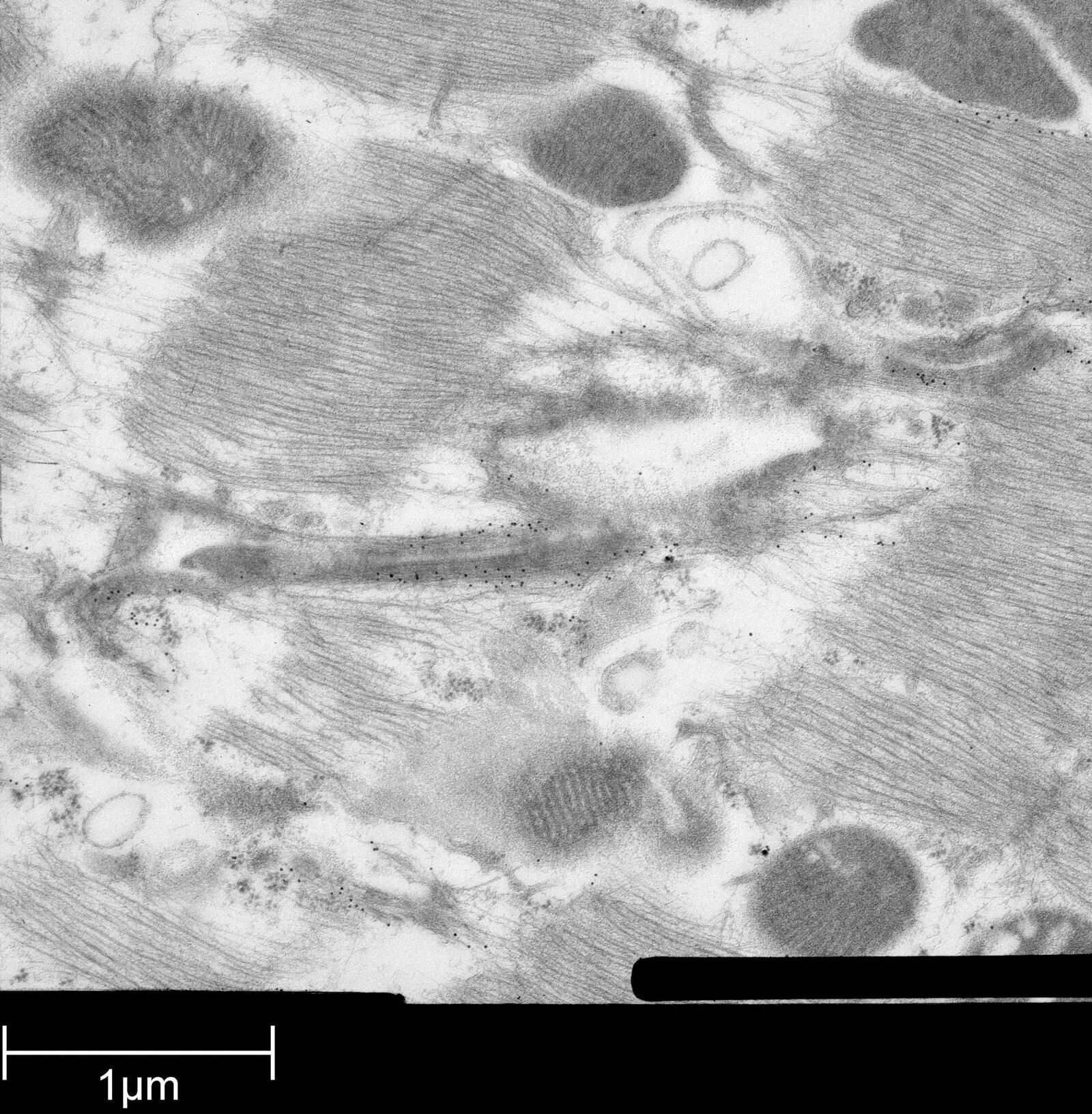
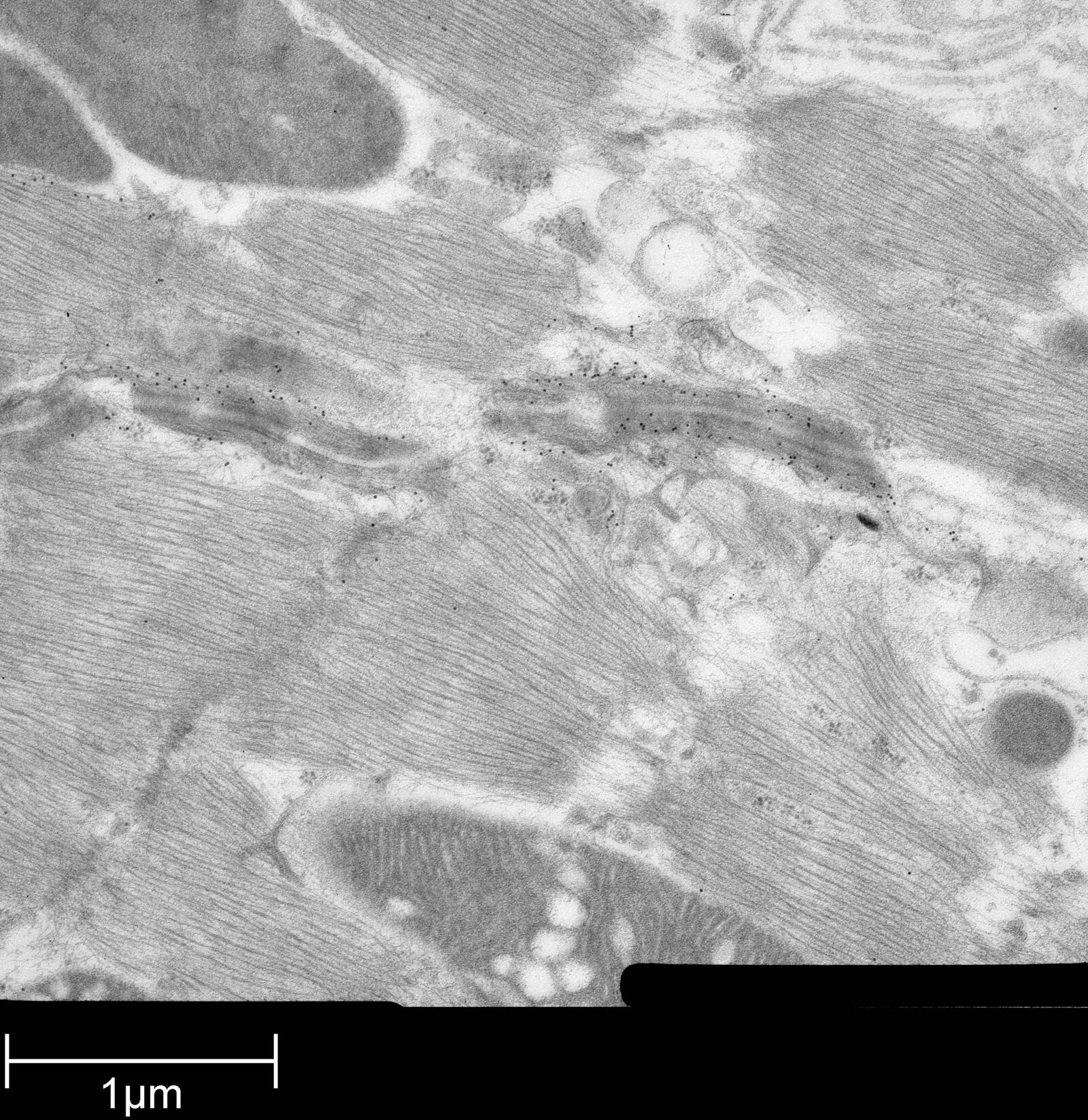
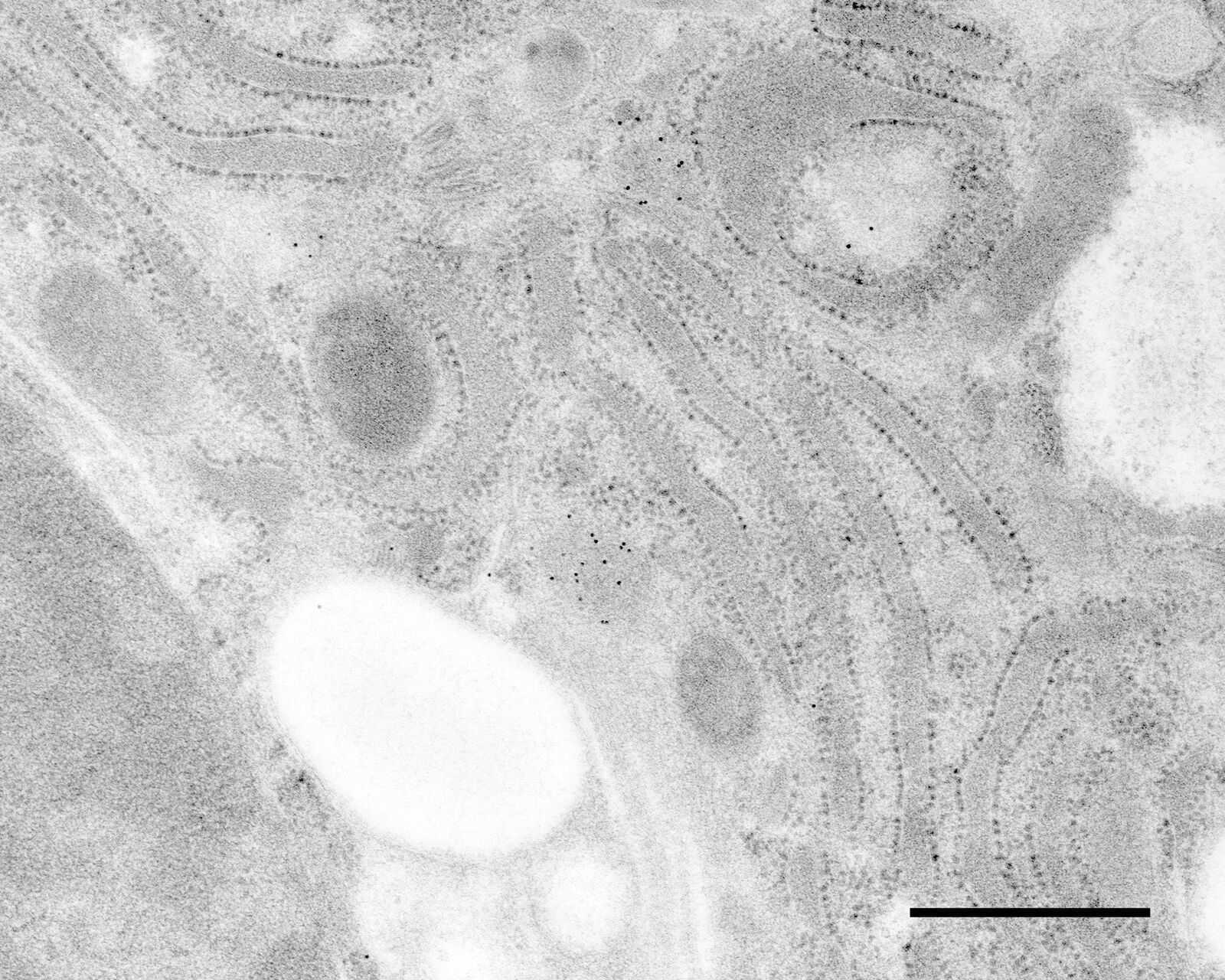
Immuno-electron microscopy of high pressure frozen and freeze substituted mouse heart
Courtesy: de Rycke R
Protocol HPF – AFS mouse heart IEM
Mouse heart tissue from wild-type (WT) and αT-catenin KO (KO) mice was excised, immersed in 20 % (w/v) BSA and frozen immediately in a high-pressure freezer (Leica EM PACT). Freeze substitution was carried out using a Leica EM AFS in dry acetone containing 2 % ddH2O, and 0.1 % glutaraldehyde over a 4-days period as follows:
- –90 °C for 24 hours,
- 2 °C per hour increase for 15 hours,
- –60 °C for 24 hours,
- 2 °C per hour increase for 15 hours, and
- –30 °C for 24 hours.
Samples were then washed 3 times in pure acetone between –30 °C and 0 °C and slowly warmed up to 4 °C, infiltrated stepwise over 3 days at 4 °C in LR-White and embedded in capsules. The polymerization was performed in Leica EM AFS using UV lamp over 6 days starting at 20 °C and ending at 37 °C.
Ultrathin sections were made using an ultramicrotome (Leica EM UC6) and post-stained in in a Leica EM AC20 for 40 min in uranyl acetate at 20 °C and for 10 min in lead citrate at 20 °C.
Grids were viewed with a JEM 1010 transmission electron microscope (JEOL, Tokyo, Japan) operating at 80 kV using Image Plate Technology from Ditabis (Pforzheim, Germany).
Immunolabeling and label quantification were performed as described previously [6].
The following primary antibodies were used for immuno-EM:
- Cx43-polyclonal rabbit (1:50; Sigma) and
- Desmin polyclonal rabbit (1:50; AbCam).
In the article of Li et al. [7] also other images are shown, processed (with Leica EM PACT and Leica EM AFS) for spur'‘s resin and HM20 (with Leica HPM010), but that's no problem, the protocol we explain here is for the shown images.
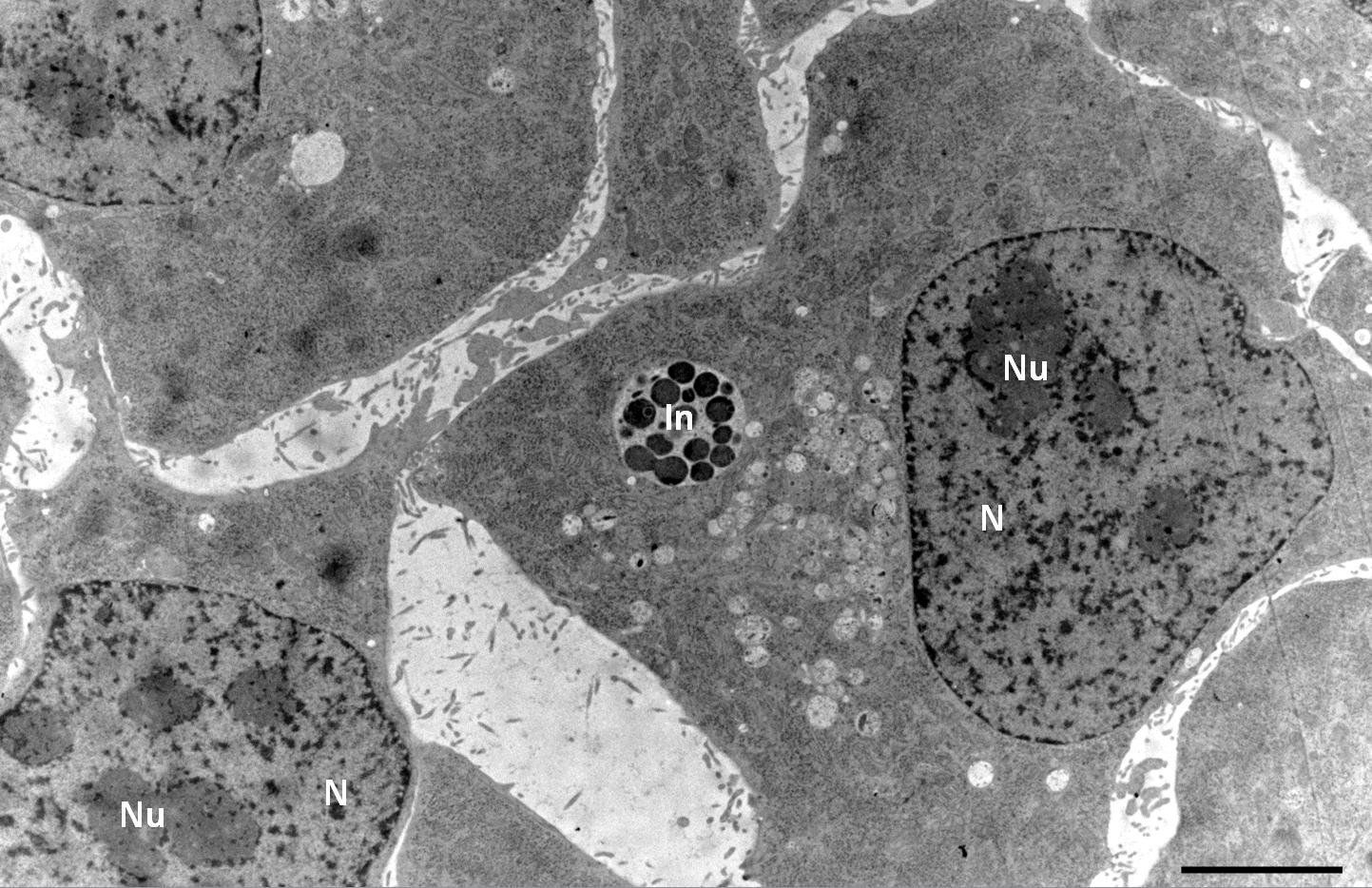
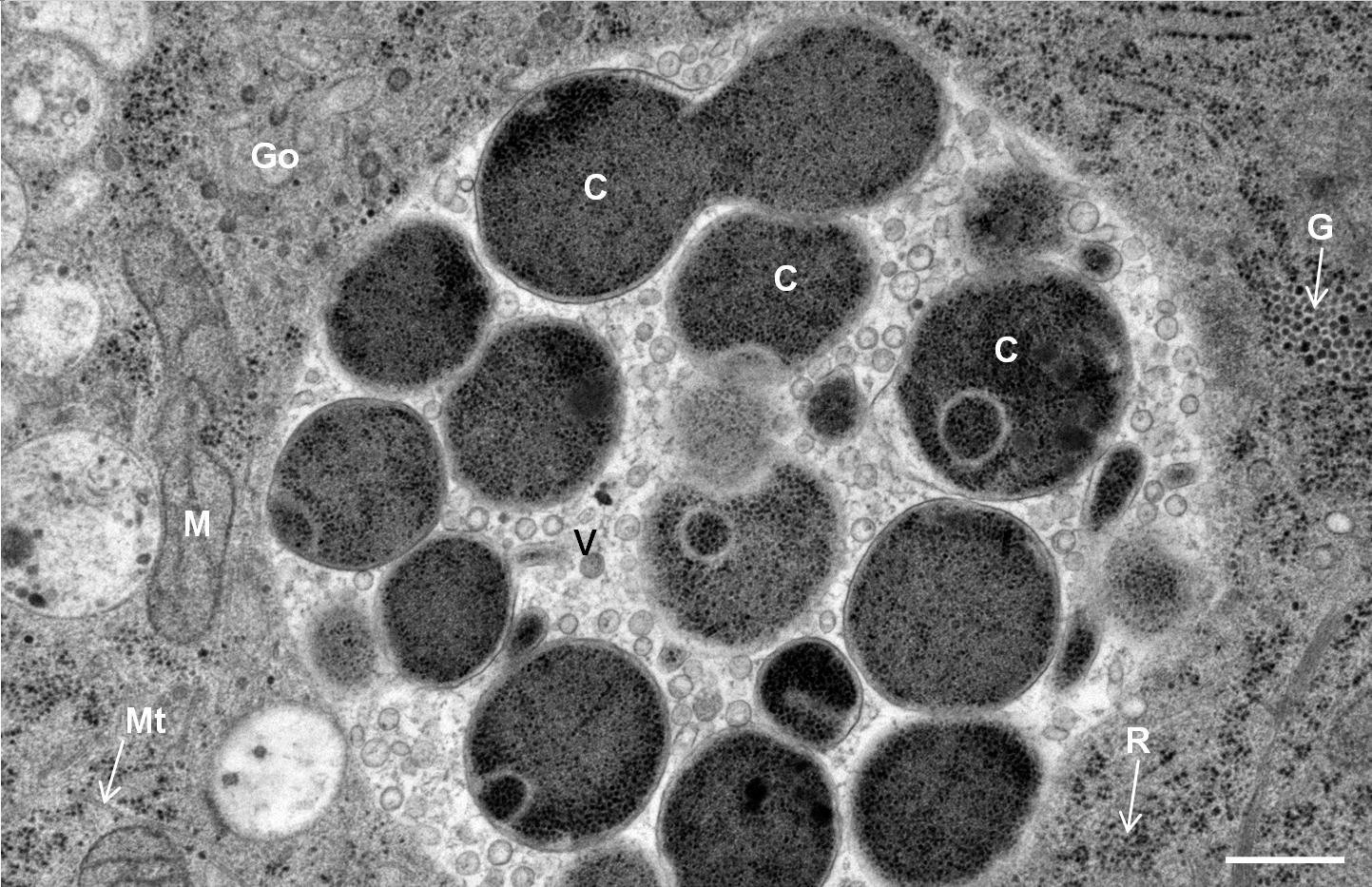
Hep-2 cells infected with Chlamydia pneumoniae
Courtesy: Kaech A
Protocol
Hep-2 cells infected with Chlamydia pneumoniae were cultured on carbon-coated 6 mm Sapphire discs. Cells were high-pressure frozen in an Leica EM HPM100 using the 6 mm CLEM middle plate with following setup: Sapphire disc with cells, spacer 200 µm, bare Sapphire disc, 2 spacers 200 µm. Ethanol was used as a synchronization fluid to transfer pressure at room temperature prior to cooling. After freezing, the Sapphire disc was removed from the middle plate in anhydrous acetone at –90 °C and immediately transferred into a 2 ml Eppendorf tube containing 2 % OsO4 in anhydrous acetone, precooled to –90 °C in an Leica EM AFS2 freeze-substitution unit.
Samples were substituted for
- 8 h at –90 °C,
- 8 h at -60 °C,
- 8 h at -30 °C, and
- 1 h at 0 °C
with periodic temperature transition gradients of 30 °C/h. Samples were then washed twice with anhydrous acetone at 4 °C, immersed in 33 % Epon/Araldite in anhydrous acetone at 4 °C overnight, followed by 66 % Epon/Araldite in anhydrous acetone at 4 °C for 6 h, and finally in 100 % Epon/Araldite at room temperature for 2 h prior to polymerization at 60 °C for at least 24 h in a 1.5 ml Eppendorf tube. Sections were post-stained with uranyl acetate and lead citrate.
NOTE: The use of ethanol as a synchronization fluid during high-pressure freezing is not necessary for cell culture monolayers on Sapphire discs. A simplified sandwich configuration can be used to provide similar results by placing the Sapphire disc in a CLEM middle plate with cells facing up covered with an aluminium specimen carrier dipped in 1-hexadecene with the 100 µm cavity facing the cells.
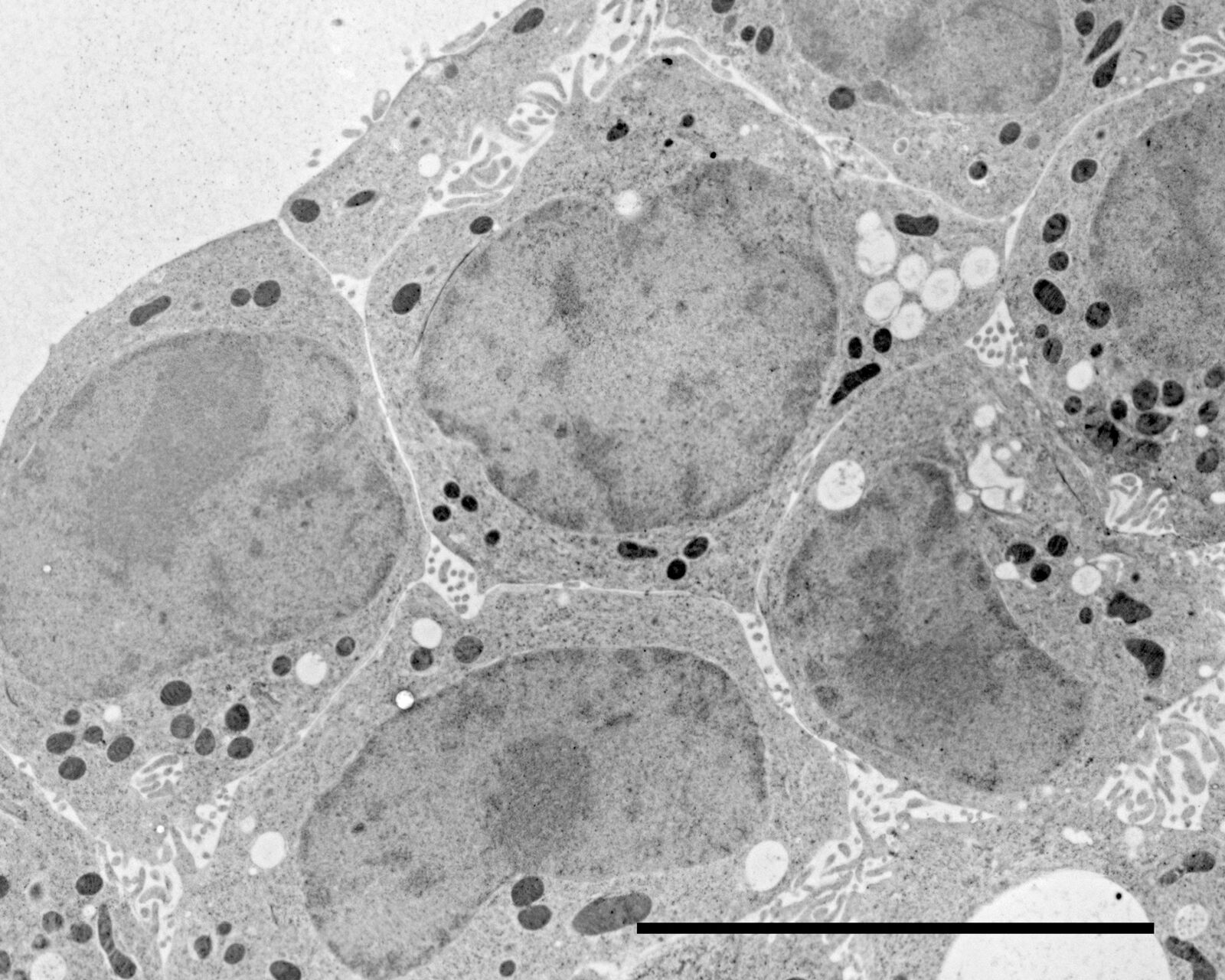
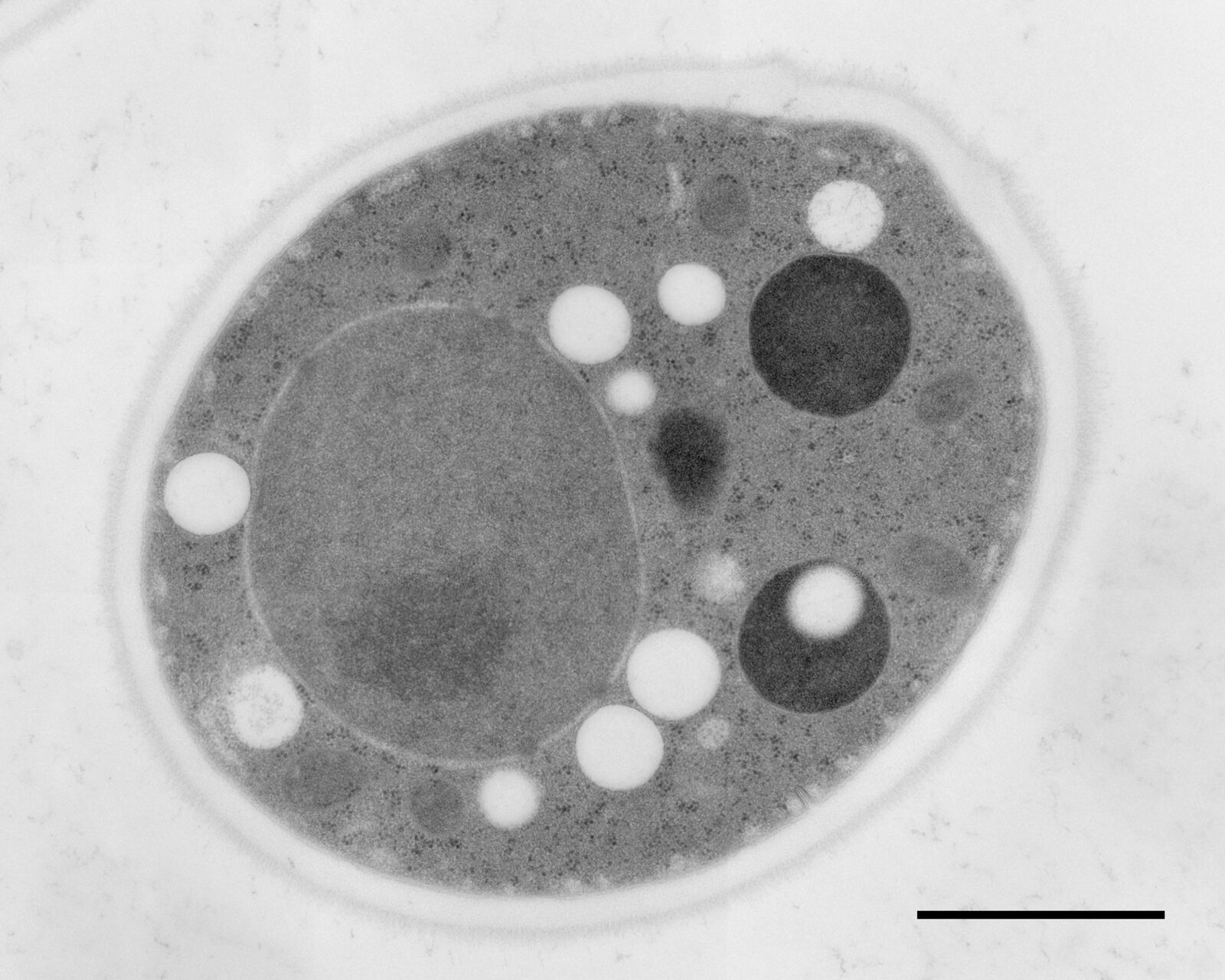
More application images
Courtesy: van Donselaar EG, Humbel BM, Utrecht University, The Netherlands; Slot JW, University Medical Center, Utrecht, The Netherlands
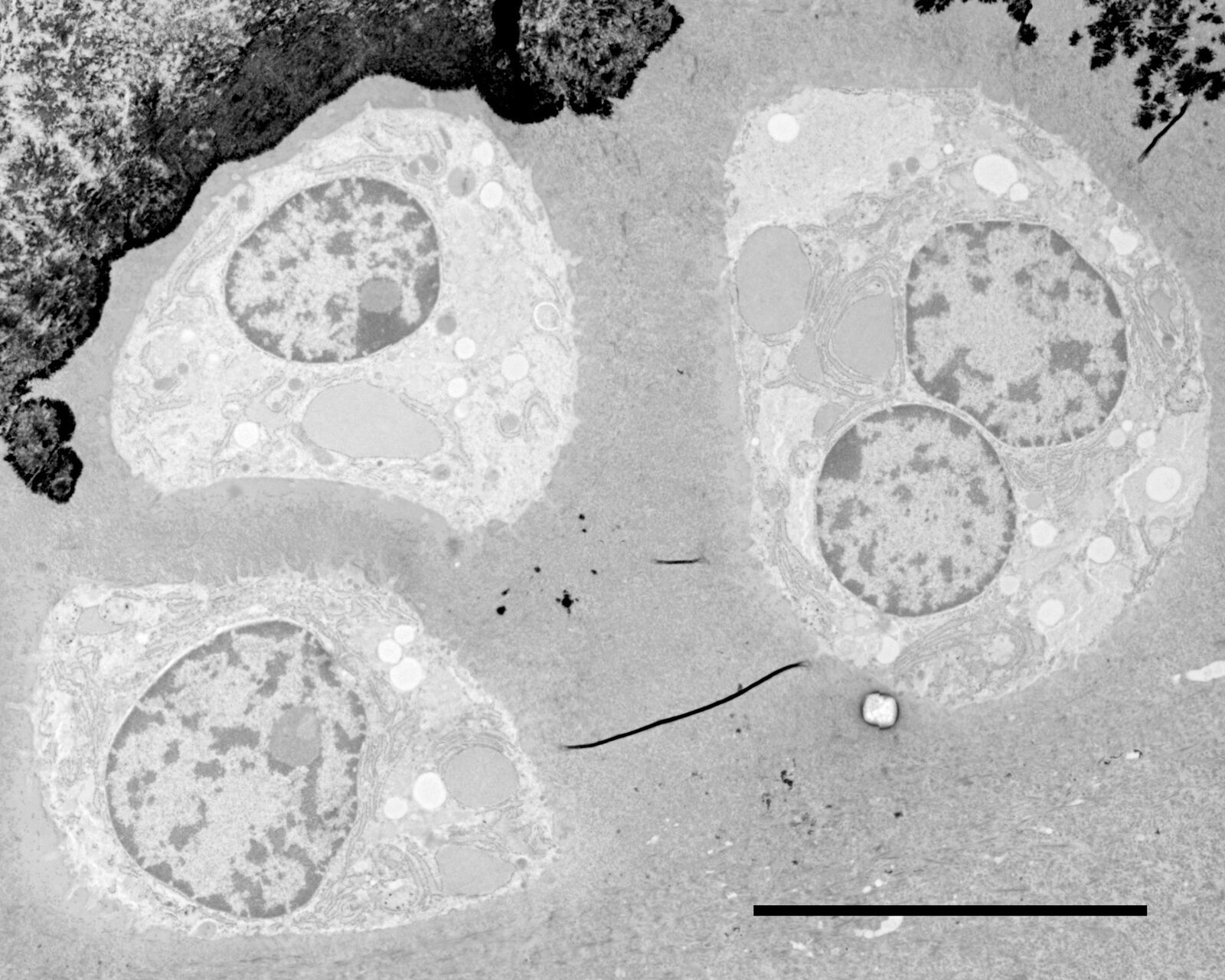
Figs. 12–14: Mouse cartilage
References
- Steinbrecht RA, Müller M: Freeze-substitution and freeze-drying. Cryotechniques in biological electron microscopy. Springer, Berlin, pp. 149–72 (1987).
- Müller M: The integrating power of cryofixation-based electron microscopy in biology. Acta Microsc. 1: 37–44 (1992).
- Hobot JA, Villiger W, Escaig J, Maeder M, Ryter A, Kellenberger E: The shape and fine structure of the nucleoid observed on sections of ultra rapid frozen and cryosubstituted bacteria. J. Bacteriol. 162: 960–71 (1985).
- Kellenberger E: The potential of cryofixation and freeze substitution: observations and theoretical considerations. J. Microsc. 163: 183–203 (1991).
- Feraru E, Feraru MI, Asaoka R, Paciorek T, de Rycke R, Tanaka H, Nakano A and Frimla J: BEX5/RabA1b Regulates trans-Golgi Network-to-Plasma Membrane Protein Trafficking in Arabidopsis. The Plant Cell 24: 1–14 (2012).
- Goossens S, Janssens B, Bonne S, de Rycke R, Braet F, van Hengel J, van Roy F: A unique and specific interaction between alphaT-catenin and plakophilin-2 in the area composita, the mixed-type junctional structure of cardiac intercalated discs. J. Cell Sci. 120: 2126–36 (2007).
- Li J, Goossens S, van Hengel J, Gao E, Cheng L, Tyberghein K, Shang X, de Rycke R, van Roy F and Radice GL: Loss of αT-catenin alters the hybrid adhering junctions in the heart and leads to dilated cardiomyopathy and ventricular arrhythmia following acute ischemia. J. Cell Sci. 15; 125 (4): 1058–67 (2012); doi: 10.1242/jcs.098640.

-b-poly(isoprene). Right: Poly(styrene)-b-poly(methyl methacrylate).")

