Key Learnings
Recommendations and requirements from regulations and guidelines for electronic records and signatures concerning quality control of medical devices.
- Overview of regulations and guidelines for the USA, EU, and China
- How the regulations and guidelines are similar and different
- Current digital inspection methods and challenges meeting regulations for electronic records
For the medical device industry, you can establish a digitally enhanced inspection solution for more efficient quality control and be ready for electronic record regulations with the help of Exalta, a smart device for traceable microscopy.
Introduction
Digital management systems for quality control have become more and more popular over the last 2 decades replacing paper-based ones used in various industries, including medical devices. Digitally enhanced visual inspection of medical devices, performed with microscopy, is more efficient and delivers more consistent results than paper-based methods. However, major regulatory agencies around the world have different regulations and guidelines for electronic records and signatures related to medical device quality control than for paper records.
Currently, many medical device manufacturers may use a visual inspection approach which has a large number of manual steps. The disadvantage of these “largely manual” visual inspection methods is that they make it difficult to fulfill the recommendations and requirements of regulations concerning electronic records and signatures.
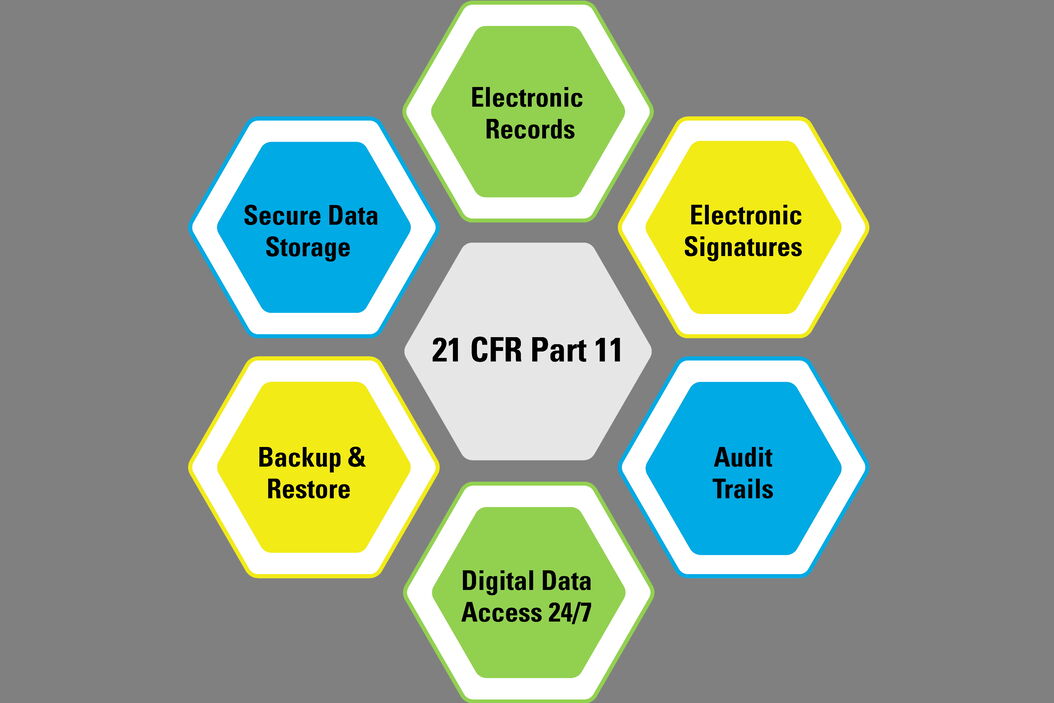
What is required by these regulations and guidelines when going digital with the quality control of medical devices? In general, the requirements for electronic records cover such things as data input, processing, recording, storage, backup, and retrieval, the reporting of results, approvals, signatures, traceability, and audit trails. Here an overview of the regulations and guidelines from the 21 CFR Part 11 for the USA [1], the GMP Annex 11 for the EU [2], and NMPA Medical Device Inspection Specifications for China [3] is provided.
Disclaimer: The overview provided in this document does not and is not intended to constitute legal advice, or any other advice, of a binding nature. All information contained in this overview is for general informational purposes only. Readers of this overview should contact their advisors or attorney to obtain advice with respect to any particular matter. Only the individual advisor or attorney can provide assurance that the information contained in this overview is applicable or appropriate to a particular situation. No representation is made that the content of the overview is complete or error-free.
It is also mentioned that a digitally enhanced visual inspection solution which minimizes manual steps throughout the entire workflow [4,5] can make it more efficient for users to meet the regulations and guidelines for electronic records and signatures medical device quality control. The Exalta smart device for traceable microscopy from Leica Microsystems helps users to establish digitally enhanced inspection [4,5] and be ready for these regulations.
Overview of Regulations and Guidelines
21 CFR Part 11 (FDA, USA)
The following is based information from the Code of Federal Regulations (CFR) Title 21, Part 11, which is published by the US Food & Drug Administration (FDA) [1]. This document gives guidance for industry concerning the scope and application of electronic records and signatures.
Narrow Interpretation of Scope
In order to promote innovation and technological advances for the benefit of public health and to avoid unnecessary controls and costs, the FDA intends to interpret the scope of part 11 narrowly [1]. This means that for records which are required to be maintained or submitted to the FDA, part 11 would apply when these records are used in electronic format rather than paper format. However, this fact does not apply to paper printouts of electronic records when the paper records already meet all the requirements.
Definition of Part 11 Records
Under this narrow interpretation, part 11 is applicable to the following electronic records [1]:
- Records that are maintained in electronic format either in place of or in addition to paper format.
- Records submitted to the FDA in electronic format.
- Electronic records which are used to perform regulated activities, in addition to documents in paper format.
- Electronic signatures which replace handwritten signatures and other ways of signing.
N.B.: It is recommended for records which need to be maintained that users determine in advance how regulated activities will be performed, i.e., whether in electronic or paper form. This decision should be clearly documented.
Validation
The decision to validate computerized systems should take into account whether users are able to meet requirements and ensure the accuracy, authenticity, and integrity of the records and signatures [1]. The FDA recommends that the approach be based on a documented risk assessment to determine how the system may affect product quality, safety, and record integrity.
Audit Trail
Compliance with all requirements regarding documentation, such as date and time, should be observed [1]. In the case there are no such requirements, audit trails or other physical, logical, or procedural security measures may still be important to ensure the trustworthiness and reliability of the records. The decision should be made based on the defined requirements, a risk assessment, record integrity, and product quality/safety.
Moreover, audit trails can be useful when creation, modification, or deletion of regulated records occurs.
Copies of Records
Appropriate access to records should be given to investigators during inspection [1]. Copies of electronic records should be supplied in common electronic formats (PDF, XML, or SGML) using established software conversion or export methods. The copying process used should produce copies that preserve the content and meaning of the record. Inspection, review, and copying of records in a form that is readable by a person should be allowed on the organization’s site using its hardware.
Record Retention
The decision of how to maintain records should be based on defined requirements and a documented risk assessment, including a determination of the record’s value over time [1]. It is important that any copies of the required records should preserve their content and meaning. Records can be archived in either non-electronic formats, such as on microfilm, microfiche, and paper, or in a standard electronic file format (e.g. PDF, XML, or SGML). Moreover, electronic and non-electronic records and signatures can co-exist, i.e., a hybrid situation.
GMP Annex 11 (EU)
The following is based on information from the EudraLex, Volume 4, Annex 11 which is published by the European Compliance Academy (ECA) Foundation [2]. This document gives guidelines for computerized systems used for good manufacturing practice (GMP) concerning medicinal products for human and veterinary use.
Principle
This annex acts as a guide to interpret the principles of good manufacturing practice (GMP) regulated activities for medicinal products [2]. It concerns all forms of computerized systems, i.e., software and hardware working together to give specific functionalities. It is specified that the use of a computerized system in place of a manual operation should not lead to a decrease in process control, quality assurance, and product quality, nor an increase in the process risk.
Validation
Documents related to validation should cover relevant life cycle steps and should include an updated listing and inventory of the systems used, including records of any change observed during the validation process [2]. The functions of the computer system should be described by the user-requirement specifications which are based on risk assessment and GMP impact. A formal assessment and reporting method should be developed to ensure quality and performance measures and include a demonstration of test methods. It should also take into account system parameter limits, data limits, and error handling. Documented assessments of testing tools and checks of data transfer should be put in place to ensure their adequacy and accuracy during this process.
Data Entry
Electronic data exchange between computerized systems and other systems should incorporate appropriate built-in checks to ensure correct and secure data entry and processing[2]. When critical data is entered manually, additional checks, e.g., by a second operator or validated electronic means, should be performed to ensure accuracy of the data. Risk management should cover the consequences of erroneous or incorrectly entered data.
Data Storage
The accessibility, readability, and accuracy of the stored data should be checked and verified during the retention period[2]. Backups should be performed regularly and the integrity, accuracy, and ability to restore the data should be monitored periodically. The ability to generate clear printouts of electronically stored data should be confirmed and batch release records should indicate any change to the original data entry.
Audit Trails
The recording of all GMP-relevant changes and deletions in an in-built system for the purposes of an audit trail should be considered and the reason for these changes should be documented [2]. Such audit trails need to be routinely available and regularly reviewed.
Electronic Signatures
Electronic signatures can be used in place of hand-written signatures for the signing of records within the organization [2]. They should include the time and date of application and be permanently linked to their respective records.
Security
The identity of operators entering, changing, confirming, or deleting data (including the date and time), records, and documents should be recorded in specially designed management systems [2].
Archiving
The archiving of data should be possible. The data should be checked for readability, accessibility, and integrity [2]. Data retrieval should be made possible when changes are made to the system, e.g., computer equipment or programs.
Medical Device Inspection Specifications (NMPA, China)
According to the China National Products Administration (NMPA) regulations concerning medical device inspection work specifications [3], the regulatory affairs and quality assurance (RA/QA) division of an organization producing medical devices can establish quality control (QC) with an electronic data and information management system for the collection, processing, recording, reporting, storage, or retrieval of QC data. In general, the NMPA uses the same requirements for electronic records as the 21 CFR part 11 [1] which has been already described above.
How are the Regulations and Guidelines Similar and Different?
As described above, the 3 regulations provide guidance regarding the use of electronic records and signatures which are applicable for medical device quality control. The NMPA regulations for electronic records follow those of FDA Part 11. The similarities and differences between Part 11 and EU Annex 11 are discussed below. A comparison overview is also given in table 1.
Similarities
- The Part 11 and Annex 11 regulations/guidelines have similar recommendations in terms of the validation, entry, storage, and archiving of electronic records and signatures.
- Both Part 11 and Annex 11 have similar recommendations about audit trails for electronic records and signatures.
Differences
- Part 11 applies to electronic records for innovation and technology concerning public health, food, and medical devices.
- Annex 11 deals with all good manufacturing practice (GMP) regulated activities for medicinal products for both human and veterinary use.
- Part 11 relates only to the maintenance, distribution and use of records and signatures in electronic format. However, Annex 11 deals with all forms of computerized systems, i.e., software and hardware working together to give specific functionalities, which are used to replace manual operations. Therefore, Part 11 has a narrower scope than Annex 11.
- Part 11 is more specific about the way electronic records should be used, saved, archived, etc. For example, it mentions the specific file formats, such as PDF, XML, or SQML, unlike Annex 11.
- Part 11 heavily emphasizes the importance of following defined requirements, whereas Annex 11 does not. Moreover Part 11 applies only to records which are required to be maintained according to the regulations.
- The use of electronic signatures is not exactly the same in the different regulations. In Annex 11 electronic signatures have the same impact as hand-written signatures within the boundaries of the company, whereas Part 11 applies to electronic signatures required by the regulations.
- Part 11 mentions the possibility to keep and use both electronic and paper records (a hybrid situation) and gives some guidelines on how to handle this situation. Annex 11 does not address a hybrid situation.
- Annex 11 emphasizes the importance of maintaining product quality, process control, and decreased risk when using computerized systems in place of manual operations. Part 11 is intended to permit the widest use of electronic technology while protecting public health.
| Requirements, Activities, and Records’ Formats | Regulations / Guidelines | |
|---|---|---|
| FDA Part 11 | EU Annex 11 | |
| Applies to all forms of computerized systems? | No | Yes |
| Applies only to electronic records and signatures? | Yes | No |
| Applies only to regulated activities? | Yes | No |
| Maintenance of electronic records with or without paper | Yes | Yes |
| Submission of electronic records with or without paper | Yes | Yes |
| Distribution of electronic records | Yes | Yes |
| Electronic records for regulated activities | Yes | N/A |
| Paper printout of electronic records | No | Yes |
| Electronic signatures in place of handwritten signing | Yes | Yes |
| Validation of computerized systems | ||
| Yes | Yes |
| Yes | Yes |
| Audit trails, or security measures | Yes | Yes |
| Copies of records | Yes | Yes |
| Electronic file formats | PDF, XML, SGML | N/A |
| Retention of records in electronic format | Yes | Yes |
| Retention of records in non-electronic format | Yes | N/A |
| Security and recording of operator’s identity | N/A | Yes |
| Archiving | ||
| Electronic format only | No | Yes |
| Hybrid situation (both paper and electronic records) | Yes | N/A |
| Regular Backups | Yes | Yes |
Table 1: Comparison of the requirements between the FDA Part 11 and EU Annex 11 regulations which concerns electronic records and signatures for quality control of medical devices. N/A is an abbreviation for “not applicable”.
Current Digital Inspection Methods and Meeting Regulations for Electronic Records
Visual inspection approaches currently in use for medical devices tend not to have a system and software which is designed to make overall inspection practical, i.e., there are many manual steps for the examination of devices and inputting and transferring of data [4,5]. As a result, this makes it less efficient to meet the requirements of regulations and guidelines for electronic records, approvals, and signatures. Exalta, a smart device for traceable microscopy from Leica Microsystems (refer to figure 1), is now available which can help users: i) establish a digitally enhanced solution for practical microscope inspection of medical devices which minimizes manual steps [4,5] and ii) be ready for regulations, like 21 CFR Part 11 and GMP Annex 11, as a result of easy record traceability, electronic signatures and approvals, rapid user authentication, and a practical audit trail.

Summary
This report gives an overview of regulations regarding the use of electronic records for medical device inspection for manufacturers in the USA (FDA 21 CFR Part 11), EU (GMP Annex 11), and China (MNPA). Digitally enhanced inspection has significant advantages over paper-based methods. These advantages lead to more consistent and efficient inspection. However, the regulations for electronic records have different requirements than those for paper ones and can vary from one country to another. At present, visual inspection methods used by manufacturers can have many manual steps, making it less efficient to follow these electronic record regulations. The Exalta smart device for traceable microscopy from Leica Microsystems enables users to establish a digitally enhanced inspection solution for more efficient medical device quality control and helps them to be ready for such electronic record regulations.
Download the article as pdf
References
- FDA Guidance Document, Part 11, Electronic Records; Electronic Signatures - Scope and Application: Guidance for Industry, September 2003, Docket Number: FDA-2003-D-0143, Food and Drug Administration (FDA).
- EC-GMP Guidelines, EU GMP Annex 11: Computerised Systems: Supplementary guidelines with specific requirements for computerized systems, June 2011, European Commission (EC), Enterprise and Industry.
- National Products Administration (NMPA) of China, Medical Device Inspection Work Specifications, Sep 2019, No. 41, index number FGWJ-2020-1740.
- J. DeRose, D.R. Barbero, Why is Manual Visual Inspection of Medical Devices so Challenging? How the challenges can be overcome with a digitally enhanced solution, Science Lab (2021) Leica Microsystems.
- J. DeRose, Going Digital for Medical Device Visual Inspection: Challenges with paper-based documentation and reporting, Science Lab (2021) Leica Microsystems.
Related Articles
-

Automotive Part Verification and Development according to Specifications
Automotive part verification during the development and production of parts and components by…
Feb 20, 2025Read article -

Visualizing Photoresist Residue and Organic Contamination on Wafers
As the scale of integrated circuits (ICs) on semiconductors passes below 10 nm, efficient detection…
Jun 12, 2024Read article -
 at the edge of a battery electrode acquired with a DVM6 digital microscope.")
Burr Detection During Battery Manufacturing
See how optical microscopy can be used for burr detection on battery electrodes and determination of…
Apr 04, 2024Read article
