
DNA double strand break induced gene editing
Since the discovery of DNA in the 1950s, researchers aim to understand this universal code of life. On the one hand, basic science wants to decipher fundamental phenomena and mechanisms. On the other hand, the applied sciences, such as biotechnology or medicine, try to use this information to create more robust crops or cure disease.
A common method of all researchers interested in DNA and the associated phenotype is modification. To find out the role of a certain gene and its respective proteins (via gene expression), an obvious strategy is to get rid of it (knock-out) or modify it (mutation) and watch the result. This goal is why over the years several techniques were developed to alter genome information.
The use of bacterial derived restriction enzymes (EcoRI, BamHI, HindIII etc.) to cut DNA at specific sites is one example of a gene altering method. These enzymes are used for molecular cloning, i.e., to fuse recombinant DNA for expression in a host organism. Each restriction enzyme has only one specific DNA sequence it can cut. Ligases fuse the targeted DNA piece into a plasmid vector. The cloning process takes place in vitro and the recombinant DNA plasmid has to be transferred back into the cell to produce the relevant protein.
A way to manipulate genes in their endogenous state, meaning directly in the target cell’s inherent genome, was developed due to the discovery of homologous recombination (HR). Exogenous DNA fragments can be introduced to the target genome by making use of sequence homology. In a few cases, a DNA fragment showing homology to the target sequence can integrate into the genome by recombination (1 in 106-109 cells). This discovery facilitated the creation of such things as knock-in (one or more genes added) and knock-out animals.
Despite these advances, the rare event of homologous recombination called for improvement. Luckily further ways to manipulate DNA came to light. It was discovered that another type of nucleic acid cleaving enzymes – nucleases – could be programmed and used for genetic manipulation. At first three major protein classes were exploited. Meganucleases derived from bacteria, zinc finger nucleases (ZFN) based on eukaryotic transcription factors, and transcription activator-like effector nucleases (TALENs) from bacteria all recognize their DNA binding site by protein-DNA interaction. Meganucleases have a DNA binding domain and a nuclease domain per se, whereas ZFs and TALEs are attached to a FokI nuclease domain.
In principle, genome editing techniques based on meganucleases, ZFN, or TALENs also use homologous recombination to introduce a piece of exogenous DNA, but they don't wait for a double strand break to occur by accident. Due to their sequence specificity and nuclease activity, they are able to produce targeted DNA double strand breaks (DSB) creating space for locus-specific homologous recombination.
Now going into more detail, there are two mechanisms which can occur after a DNA double strand is cleaved. Both are part of the endogenous DNA repair machinery and depend on the presence of an adequate homology repair template (see Fig. 1):
A) Non-homologous end-joining (NHEJ) happens in the absence of an exogenous homology repair template and can introduce insertions or deletions (indels) eventually leading to frameshift mutations or gene knockouts.
B) Homology-directed repair (HDR) happens if an exogenous repair template (for example, a manipulated gene) is available and can be inserted into the opened DNA strand by the repair machinery producing a precise gene modification.

The CRISPR/Cas bacterial immune system
The CRISPR/Cas system was discovered first in the 1980s, when researchers took a closer look at the genome of bacteria. Initially studying certain enzymes, they discovered repetitive elements downstream from their target gene’s sequence. These repeat elements in turn were interspaced by non-repetitive sequences. With the sequencing of more and more genomes, it turned out that these interspaced repeats were conserved throughout the genome of plenty of bacteria and archaea, suggesting that this might be something important. Due to their characteristics they were named Clustered Regularly Interspaced Short Plindromic Repeats or CRISPR.
The next decisive discovery was made in 2005, when the analysis of the spacers between the repetitive elements revealed their extra-bacterial origin. Instead, they belonged to the genome of bacteriophages (viruses). Then an interesting revelation occurred: bacteria which were carrying DNA elements of certain phages could no longer be infected by them. This result led to the conclusion that the newly discovered genetic elements might belong to a sophisticated prokaryotic immune system (see Figure 2).

Subsequent studies proved this theory right and brought to light that there are several different CRISPR based systems which in the end all lead to the degradation of viral DNA respectively RNA. The basic principle of CRISPR is to integrate segments of foreign DNA into the bacteria genome and use their transcripts as a matrix for base pairing induced binding and destruction of the viral DNA for all future infections. A more detailed look at the CRISPR mechanism is described below.
Infection of the bacterial cell starts with a phage, docking at the surface, releasing its genetic material into the bacterium (see Fig. 3). At the same time, some genes upstream of the CRISPR locus are expressed. Due to their location and function they were named CRISPR associated genes or in short Cas. Their gene products are able to cut out distinct segments from the foreign DNA, which are called Protospacers. Subsequently, a protospacer is inserted as a DNA sequence spacer into the CRISPR locus.
Transcription of the CRISPR locus leads to a pre-crRNA composed of many spacers and repeats, which is processed later on. The mature crRNA then contains only one repeat sequence and one spacer and can be utilized by different CRISPR/Cas systems for targeting foreign DNA via Watson-Crick base pairing. The type I, III, and IV systems consist of large complexes of Cas proteins, whereas the type II (i.e. Cas9) and type V (i.e. Cpf1) systems only consist of a single protein for both DNA targeting and cleavage. In addition to the crRNA, the Cas9 system is comprised of another short segment of RNA, called tracrRNA (trans-activating crRNA), which hybridizes with the spacer part of the crRNA.
The end result of all CRISPR/Cas systems is the same: after crRNA guides the binding of the Cas proteins to the homolog segment, the foreign DNA is degraded. The fact that the spacers inside the bacterial CRISPR locus are comprised of the same sequences as the viral DNA raises the question: why don’t the Cas proteins recognize the spacers in the bacterial DNA?
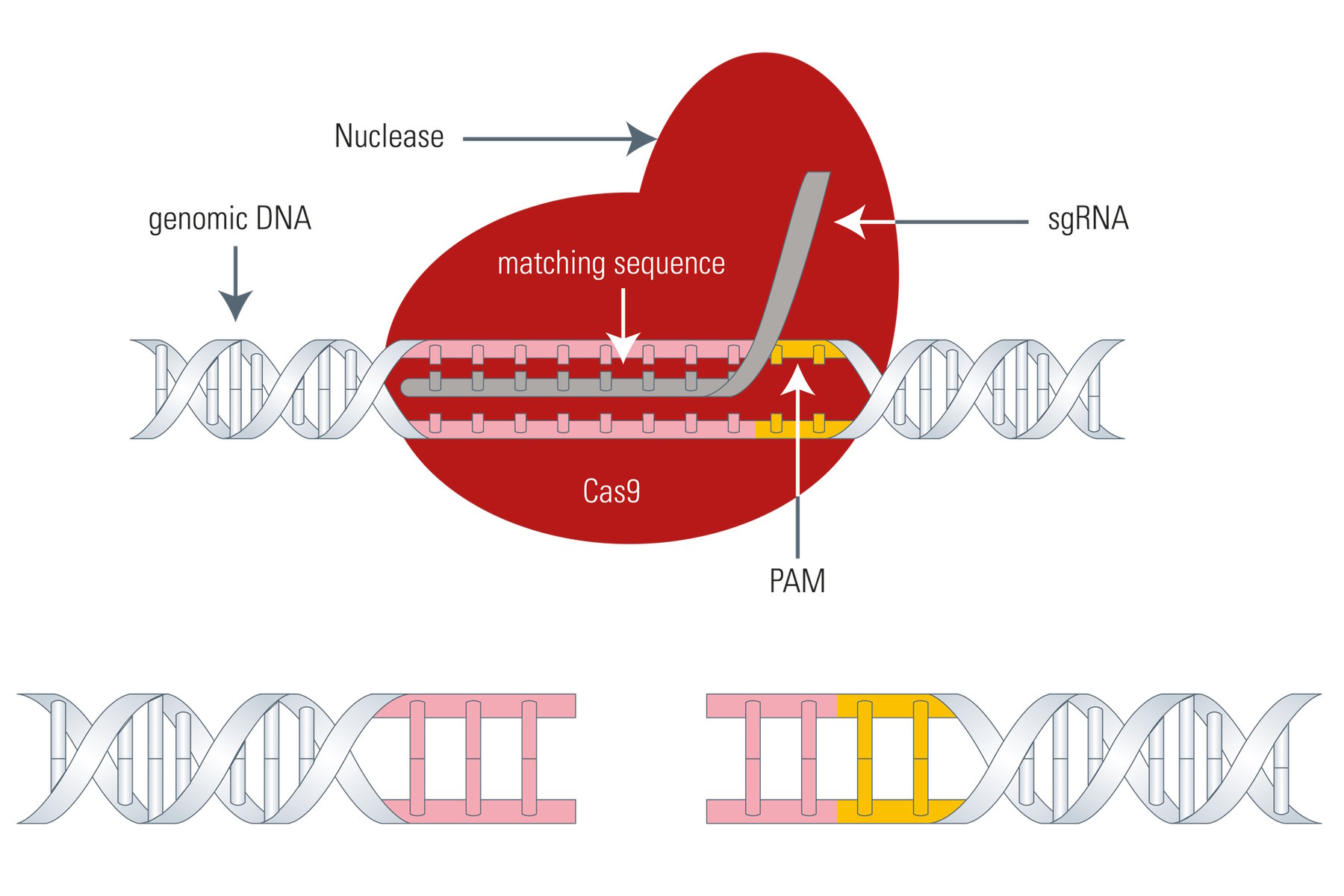
It is curious that for systems I and II there is a mechanism to discriminate between self and non-self. The Cas proteins need a so called PAM (Protospacer- adjacent motif) to be able to function effectively once bound with the viral DNA strand. Depending on the organism, the PAM can differ in its sequence. One PAM derived from Streptococcus pyogenes is 5'-NGG-3', where "N" is any nucleobase and "G" is guanine. Because the direct repeats in the CRISPR locus flanking the acquired viral gene excerpts (spacers) do not have a PAM, the Cas9 protein won’t bind there. It only binds to invading viral DNA due to infection. In other words, with help from the PAM, the CRIPSR locus is protected from self-destruction.

Gene editing with Cas9
The Cas9 nuclease from the bacterial type II CRISPR/Cas system can be utilized for genome editing. That means it is able to introduce exogenous genes or disturb endogenous ones. With the help of such experiments, researchers can discover the role of certain genes and their respectively expressed proteins. Moreover, they can edit genomes to modify organisms for beneficial purposes, e.g., more robust, easier-to-cultivate crops.
Very often a microscope is used in the course of such trials for specimen preparation (e.g., cell culture work) or to observe the phenotype of the genetically modified cells or organisms.
The Cas9 gene editing system follows the same principle as meganucleases, ZFNs, or TALENs: the Cas9 nuclease is able to cleave the DNA strand at a defined site and cut it to produce a double-strand break making it susceptible for the above mentioned DNA repair mechanisms (see Fig. 4). The big difference between Cas9 and the three existing genome-editing tools is that Cas9 uses a piece of RNA for DNA targeting. This fact is very advantageous, because the so called sgRNA (single guide RNA) can be synthesized very easily. Instead of designing DNA-binding proteins in a laborious manner, the sgRNA can be synthesized very fast by PCR, corresponding to the host DNA sequence.
The sgRNA goes back to the crRNA and tracrRNA of the original CRISPR/Cas9 system. For simplification, crRNA and tracrRNA were fused by the introduction of a loop, resulting in sgRNA.
However, the Cas9 system has also a drawback. As mentioned above, the Cas9 nuclease needs a PAM (Protospacer adjacent motif) in close proximity to the target DNA sequence (3’ end) or it cannot effectively work, thus, restricting its gene editing potential to a certain degree.
So, for the utilization of Cas9 as a gene-editing tool, only DNA sequences which are flanked by a PAM at the 3’ end can be modified. As an example, the above mentioned 5'-NGG-3' PAM enables the human genome to be edited at every 8 base pairs on average with the Streptococcus pyogenes derived Cas9 (SpCas9). However, Cas9 proteins from other species, such as Streptococcus thermophilus or Neisseria meningitidis, have other PAMs which in turn expand the targeting range of the Cas9 toolbox.

The greatest interest in the Cas9 system stems from the possibility to use it for multiplex editing in mammalian cells, i.e., to edit different target sites simultaneously by adding several sgRNAs. Further advantages, as well as drawbacks, of the Cas9 system are listed in table 1 below.
| Meganuclease | Zinc Finger Nucleases | TALENs | CRISPR/Cas9 |
DNA Binding Molecule | Protein | Protein | Protein | RNA |
Origin | Bacteria | Eukaryotes | Bacteria | Bacteria/Archaea |
Pros & Cons | - Small number of suitable meganuclease protein residues fitting to target DNA sequences | - High effort to modify - Consists of a modular array of DNA-binding proteins - Crosstalk between them influences sequence specificity - Requires extensive screening | - High effort to modify - As for ZFNs, the modular construction can suffer from context-dependent specificity - Labor intensive | + Easy to modify + Fast production of sgRNA + Highly customizable + Multi targeting (Multiplexing) - Needs a PAM sequence at the target site |
Table 1: Facts, advantages and disadvantages of genome editing tools.
Application of the Cas9 system
For the delivery of the two elements – Cas9 and sgRNA – into the target cell there are diverse strategies.
Editing of cell culture lines can be done with plasmid transformation. Either both elements are coded on the same vector or two separate ones. When an insertion or replacement is planned, the donor DNA has to be transferred with an additional plasmid or single strand oligonucleotide (ssODN).
For the generation of transgenic animals, fertilized zygotes can be microinjected with a purified Cas9 protein and in-vitro-transcribed sgRNA. Somatic gene modifications can be performed with the help of viral vectors encoding the Cas9 and sgRNA.
One major advantage of the Cas9 system can be found in the generation of transgenic animals. As mentioned above, it is sufficient to transfer the sgRNA and Cas9 protein plus the required DNA directly into the zygote (fertilized egg). The Cas9 procedure can modify multiple alleles in one run. In comparison, other techniques depend on the time-consuming genetic manipulation of embryonic stem cells, prior to their injection into a blastocyst (later embryonic stage). The stem cell procedure delivers heterozygote individuals making a long mating process necessary to end up with homozygote animals. In this case, the Cas9 system can save the researcher about one year.
In addition, the Cas9 system can also be utilized with full-grown animals. This new option might boost therapeutic gene editing and give researchers new chances to cure diverse diseases.

On top of its use as a gene editing tool, the Cas9 system has a lot of other potential applications. Not only can the DNA cleavage function be exploited, but, by suppressing the nuclease activity, the Cas9 protein can be converted into a highly specific DNA probe. This function of Cas9 can fulfill a broad range of tasks. For example, the Cas9 protein can be fused to a transcription activator for targeted gene expression. When coupled with a fluorescent protein, the Cas9 protein can label distinct DNA loci allowing the study of chromosome dynamics. Moreover, chemo- and/or photo-activated Cas9 proteins have been established to gain temporal control of gene expression.
Outlook
The Cas9 nuclease for gene editing originated from the bacterial antiviral CRISPR/Cas9 system. In combination with a synthetic sgRNA, researchers can knockout or knockin genes more easily and quickly than before. This development creates new opportunities for basic research, especially for drug development or medical therapeutics. Moreover, applied sciences will have the possibility to create more lucrative crops or microorganisms to produce all kinds of basic materials.
The Cas9 system has also the potential to boost gene therapy approaches. One can imagine to directly eliminate harmful mutations which produce diseases or to excise HIV gene sequences from T-cells to avoid the onset of AIDS.
In addition to its major impact on molecular biology, numerous microscopy applications have arisen from the Cas9 nuclease, e.g., endogenous protein labelling, dynamic imaging of gene loci, or the development of photo-activated transcription systems.
References
- Mojica FJ, Díez-Villaseñor C, Soria E, Juez G (2000). "Biological significance of a family of regularly spaced repeats in the genomes of Archaea, Bacteria and mitochondria". Molecular Microbiology 36 (1): 244–6.
- Horvath P, Barrangou R (2010). "CRISPR/Cas, the immune system of bacteria and archaea". Science 327 (5962): 167–70.
- Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E (2012). "A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity". Science 337 (6096): 816–821.
- Makarova KS, Aravind L, Wolf YI, Koonin EV (2011). "Unification of Cas protein families and a simple scenario for the origin and evolution of CRISPR-Cas systems". Biology Direct 6: 38.
- Mali P, Esvelt KM, Church GM (2013). "Cas9 as a versatile tool for engineering biology". Nature Methods 10 (10): 957–63.
- Sampson TR, Saroj SD, Llewellyn AC, Tzeng YL, Weiss DS (2013). "A CRISPR/Cas system mediates bacterial innate immune evasion and virulence". Nature 497 (7448): 254–7.
- Plagens A, Richter H, Charpentier E and Randau L. (2015) DNA and RNA interference mechanisms by CRISPR-Cas surveillance complexes. Fems Microbiol Rev, 39: 442-463.
- Makarova et al. (2015) An updated evolutionary classification of CRISPR–Cas systems. Nature Reviews Microbiology, 13: 722–736.
- Zetsche B, Gootenberg J, Abudayyeh O, Slaymaker I, Makarova K, Essletzbichler P, Volz S, Joung J, van der Oost J, Regev A, Koonin EV, Zhang F (2015) Cpf1 Is a Single RNA-Guided Endonuclease of a Class 2 CRISPR-Cas System. Cell 163 (3): 759–771.