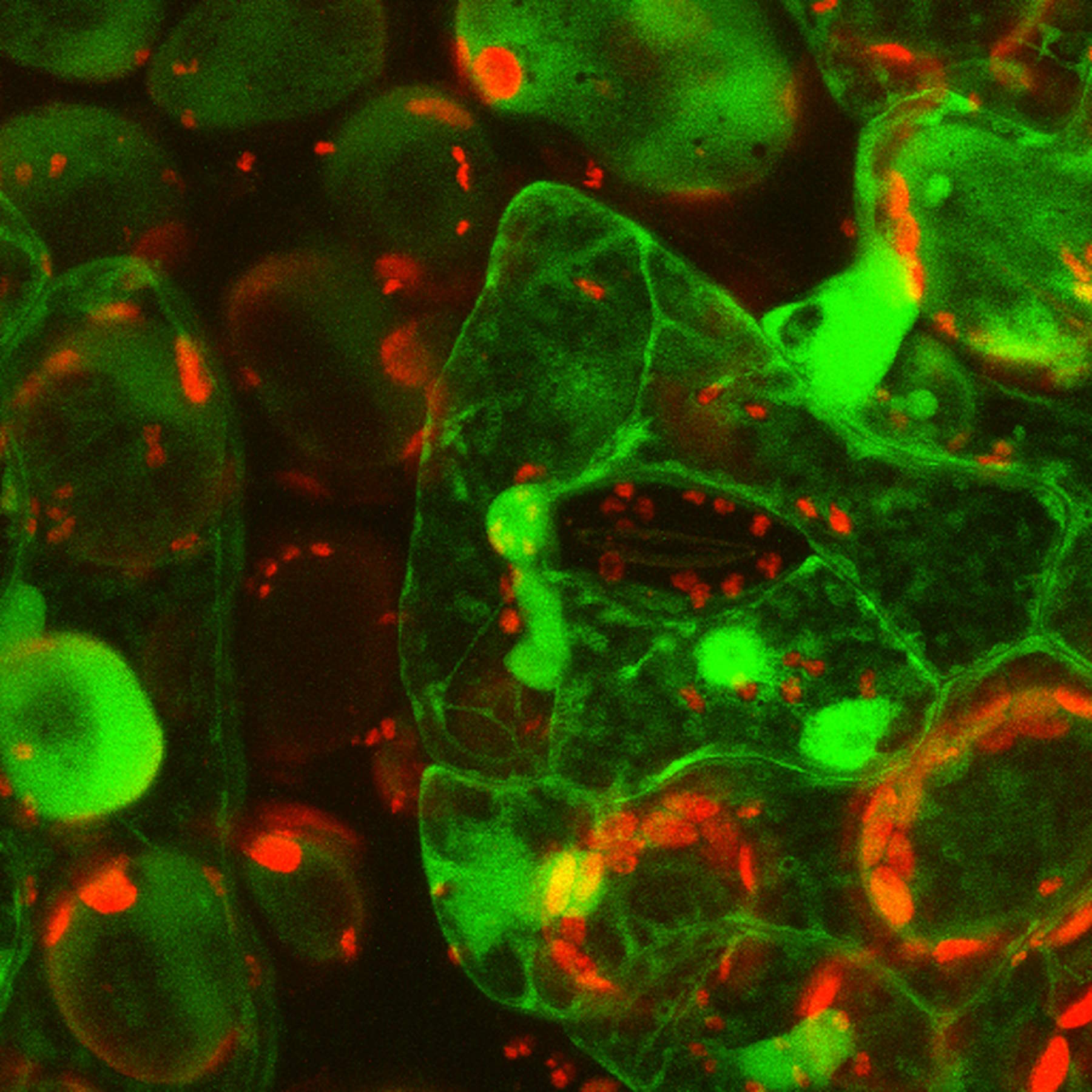
 inoculated with cowpea mosaic virus (CPMV) containing the GFP-gene inserted between the movement protein (MP) and the capsid proteins (CPs) in the viral RNA 2")
The challenge to observe and analyze living cells
The acquisition of information about the “molecular state” of a sample already is a hard task in fixed cells or tissue. This becomes even harder, if the information has to be acquired in real-time, as the cells during an experiment have to be functioning as naturally as possible. Additionally, a high amount of information has to be sampled in a relatively short time, as many events only last seconds or even milliseconds (e.g. changes in cellular ion levels).
One approach to address these challenging demands are certain optical methods that are collectively called live-cell imaging. Live-cell imaging allows for investigation of dynamical physiological processes in living cells instead of giving a “snapshot” of a cell’s current state. It turns Snapshots to movies. Live-cell imaging provides spatial and temporal information of dynamic molecular events in single cells, cellular networks (in situ) or even whole organisms (in vivo). These features make live-cell imaging a requisite technique for addressing physiological questions in cell biology, cancer research, developmental biology and neuroscience.
In recent years substantial advances in electronics, optics and biochemistry made live-cell imaging easily accessible for scientists. Using optimized microscopes, specialized light sources, high speed cameras, highly sensitive detectors and exclusively developed fluorescent markers, live-cell imaging methods nowadays provide both - a technically mature and still innovative toolsets to address the challenging demands of investigating single cells or whole cellular networks on a molecular level in real-time.
Live-cell imaging of cells with a mitochondrial marker (MitoRed®) and a fluorescent calcium dye (Fluo-4)
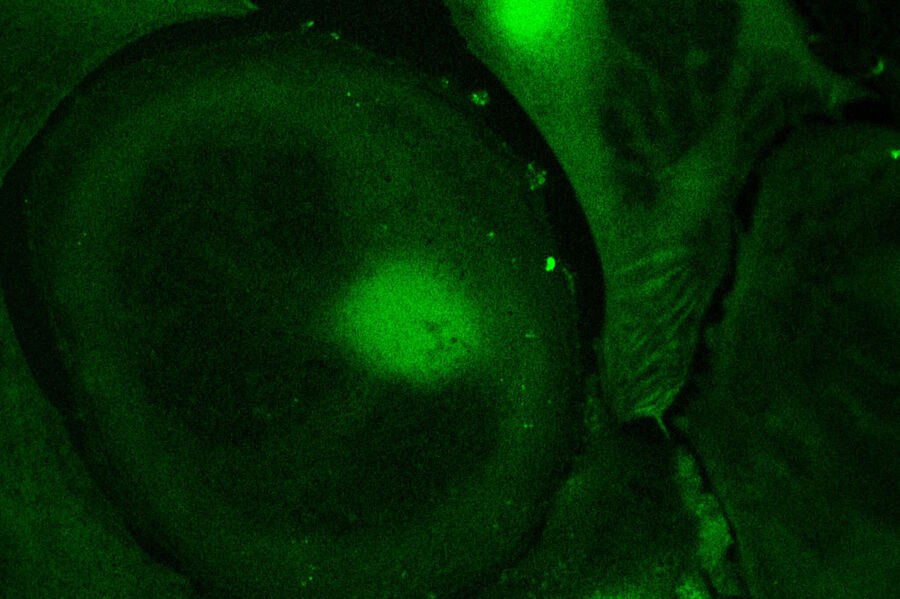
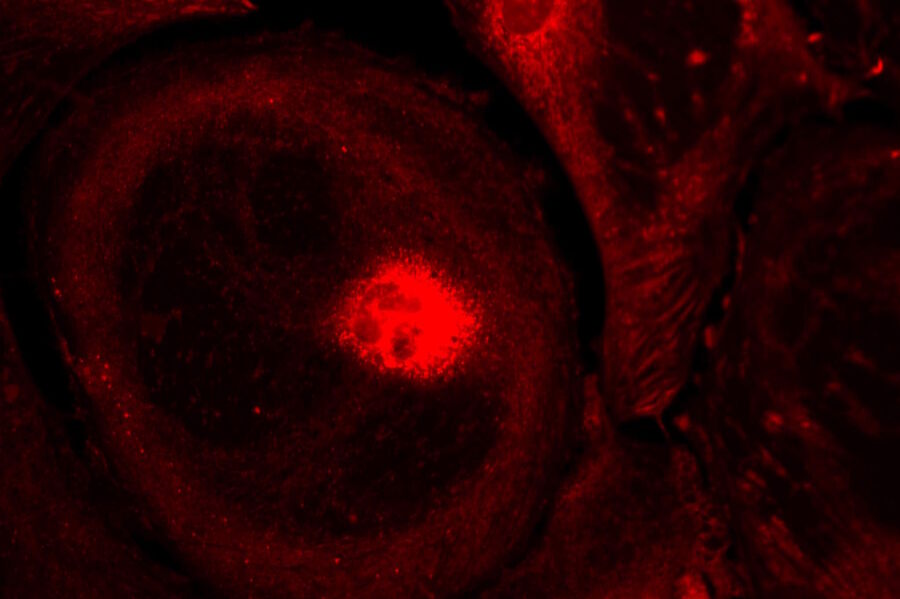
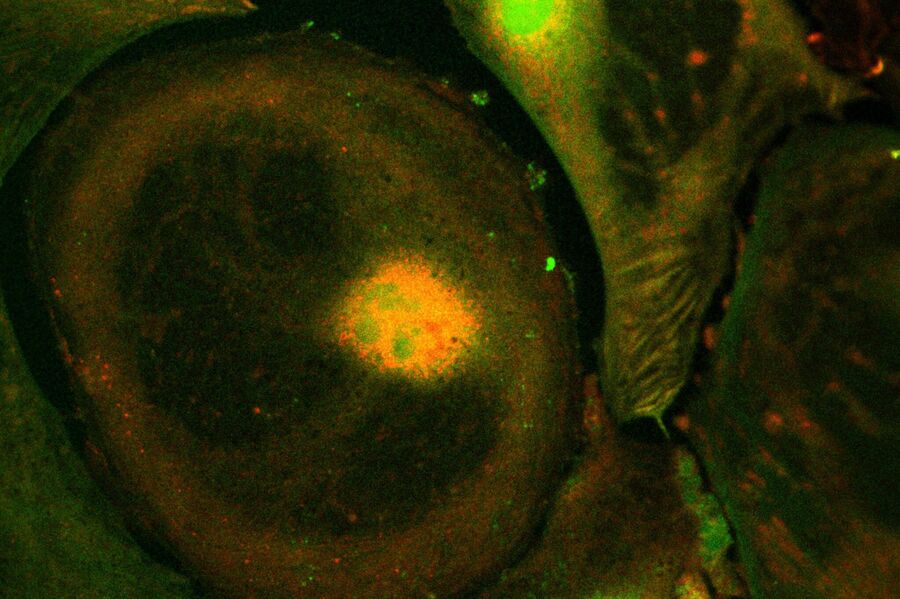
Images acquired with a confocal microscope Leica TCS SP5 (DMI6000 CFS) and Leica LAS AF7000 imaging software. Courtesy of Dr. Sophie Veitinger, RWTH Aachen University, Institut of Biology II, Department of Chemosensation, Germany. Present address: Dr. Sophie Veitinger, Philipps University Marburg, Institute of Cytobiology and Cytopathology, Germany
Corresponding publication (not the image source):
Purinergic signalling mobilizes mitochondrial Ca²⁺ in mouse Sertoli cells, Veitinger S, Veitinger T, Cainarca S, Fluegge D, Engelhardt CH, Lohmer S, Hatt H, Corazza S, Spehr J, Neuhaus EM, Spehr M; Journal of Physiology. 2011 Nov 1;589(Pt 21):5033-55
General issues in live-cell imaging
Live-cell imaging generally is possible with cultured cell lines (e.g. HEK cells, HeLa cells), primary cell cultures (e.g. skin cells, neural cells), acute slice preparations (e.g. brain slices) or whole organs or organisms. A major task is to keep the cells in a healthy state during the experiments as the cells suffer from phototoxicity and are taken out of their “natural” environment.
Extracelluar solutions
Different types of artificial extracellular solutions (Ringer’s solution, artificial cerebro-spinal fluid (ACSF) and media (e.g. Leibovitz L-15) are used to provide the cells with essential ions and other cofactors to sustain their physiological functions. The composition of media used for live-cell imaging ranges from very simple "salty" solutions (e.g, Ringer’s solution) to rather complex mixtures (e.g. Leibovitz L-15).
However, all solutions have in common that a pH buffer system is included, as exposure to the environment can dramatically change the medium’s pH (usually pH 7.2.-7.4). In many extracellular solutions pH-buffering is achieved via the addition of 10–20 mM HEPES (2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid), a zwitterionic organic chemical. Unfortunately, for many cells extracellular solutions cannot be buffered with HEPES or other chemical buffers (e.g. MOPS, TES), as a lack of pH-buffering bicarbonate in the medium harms the cells. To overcome this issue, carbon dioxide has to be delivered to the extracellular solution (carbon dioxide is turned over to bicarbonate when in contact with extracellular solution). This can be achieved in two ways: One is to constantly deliver gaseous carbon dioxide (usually in the form of carbogen: 95 % oxygen with 5 % carbon dioxide) to the extracellular solution and constantly superfuse the cells. This method is commonly used for slice preparations with a higher metabolic turnover than cell cultures.
Another common method is to keep cells in culture chambers designed for a regulated atmosphere and, in many cases, temperature. In such cultures an atmosphere of 5–7 % carbon dioxide is constantly supplied and temperature can be tightly controlled. However, to decide whether a chemically buffered extracellular solution is sufficient to keep the cells in good condition or whether it is necessary to deliver carbon dioxide or even use climate chambers, one has to consider the type of sample that is used and the duration of the experiment. In many cases a chemically buffered solution is sufficient for cell cultures and rather short experiments. Working with acute slices usually demands a sufficient carbon dioxide supply, as the metabolic turnover is much higher. However, for many cell types and longtime experiments a climate chamber is mandatory.
Phototoxicity
Another issue in live-cell imaging with fluorescence dyes, is damage caused to the cells by incident light from lasers or high-intensity arc-discharge lamps, so called phototoxicity. Phototoxicity occurs mainly if synthetic fluorescent dyes are excited. In their excited state many of them react with molecular oxygen and produce free radicals. To avoid phototoxicity, it is mandatory to choose the lowest possible light intensity and shortest possible excitation duration, to keep the dose of incident high energy light as low as possible. During experiment design, it is mandatory to also consider the duration of the experiment. In long-term experiments a high frame rate often is unnecessary. Therefore, the cycle frequency of image acquisition can often dramatically be lowered from e.g. more than 10 frames per second to 1 frame per second or even lower. This substantially lowers the dose of incident light on the sample and therefore drastically lowers phototoxicity.
One might also think about changing image acquisition settings, as in most of the cases low intensity of fluorescent signals can be improved by using camera features as binning or improved gain or by the usage of special low light cameras (e.g. EM-CCD cameras). Doing so, a better signal to noise ratio and signal quality can be achieved without increasing excitation duration or light intensity, both causing higher phototoxicity. Additionally, choosing fluorophores with long excitation wavelengths prevents from phototoxicity, as the energy delivered to the sample is low compared to fluorophores with short excitation wavelengths. Fluorescent proteins (as e.g. green fluorescent proteins (GFP)), usually are not phototoxic, as their photoactive site is deep inside the protein covered by a polypeptide envelope.
Focus drift
Furthermore the occurrence of focus drift, especially in long term experiments, can be an issue during live-cell imaging experiments. This can be avoided by using imaging setups equipped with software or hardware controlled autofocus.
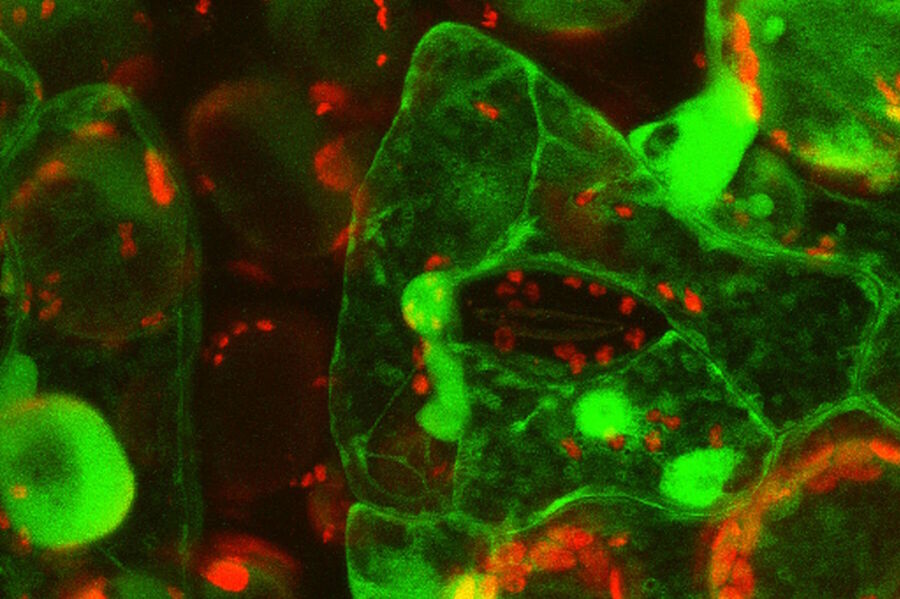
Fig. 5: Primary leaves of cowpea (Vigna unguiculata "California Blackeye") inoculated with cowpea mosaic virus (CPMV) containing the GFP-gene inserted between the movement protein (MP) and the capsid proteins (CPs) in the viral RNA 2. The GFP is expressed in abundance as a free protein (so NOT fused to either MP or CP) and can be localized in the cytoplasm and nucleus of infected epidermal and mesophyll cells. Courtesy to Dr. Joan Wellink, Dept. Biomolecular Sciences, Lab. of Molecular Biology and Dr. Jan van Lent, Dept. of Plant Plant Sciences, Lab. of Virology, Wageningen Agricultural University, The Netherlands.
Video 1: Live onion bulb cells stained with DIOC6 with simultaneous transmitted light detection; aquired with a confocal microscope Leica TCS SP2 AOBS RS, objective 63x, zoom 1.5, two fold line average, format 512 x 265, 4.7 frames per second.
Video 2: COS cells transiently transfected with a construct expressing a GFP-fused protein. The mainly cytosolic protein is forming needles in the cell. 3D stacks (10.14 µm) were recorded every 5 seconds with 14 sections/stack for 10 minutes. For the movie the maximum projections of the stacks were used. 512 x 512 pixels, bidirectional scan, Zoom 2, objective HCX APO L U-V-I 63.0 x 0.90 W UV. Courtesy of Jocelyn Laporte, Equipe Génétique Humaine, centre imagerie, IGBMC, Illkirch, France.
Methods employed for live cell imaging
The range of widefield and confocal microscopic techniques applied for live-cell imaging is also extremely wide. Often, the growth of cells, cell aggregates or cell movement is observed over time using compound microscopes and contrasting methods like phase contrast and differential interference contrast (DIC). Also, time- lapse imaging of larger specimens, e.g. developing zebrafish embryos, is widely performed, usually with stereo microscopes or macroscopes. Advanced fluorescence techniques have become more and more important in the last decades. The rapid increase in the application of confocal microscopy has changed the perspective in biological investigation from planar to the third dimension.
 and phalloidin (magenta), imaged using Viventis SCAPE; scale bar 50μm. Courtesy of Marina Cuenca and Heleen Jungen (Dayton lab), EMBL Barcelona.")


