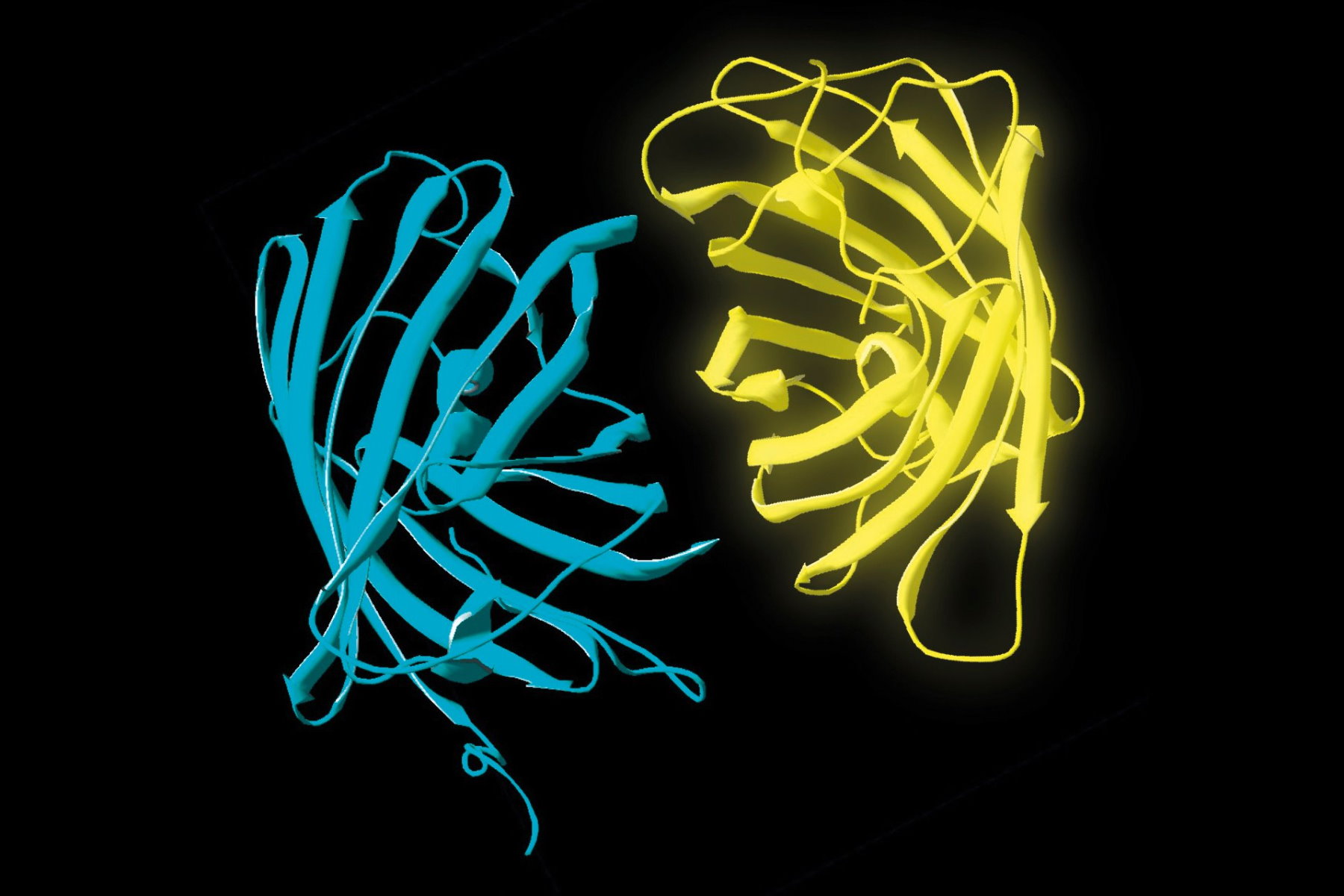
 and acceptor (A) molecule which participate in FRET (Förster resonance energy transfer).")
Förster resonance energy transfer (FRET) is a well-established fluorescence-based technique which is used to study molecular interactions. It is useful for the analysis of protein-DNA and protein-protein interactions as well as estimating intermolecular distances on a nanometer scale. When FRET is measured using fluorescence lifetime imaging microscopy (FLIM), then data quality and precision is improved. More details about the FLIM-FRET method are discussed in this article.
Motivation for using FLIM-FRET
Intensity-based FRET methods (for more about the basics of FRET, refer to the sections below), which work only when fluorescent donor and acceptor molecules are available, are quite susceptible to variations in fluorophore concentration. The advantage of FLIM-FRET is that it requires only the donor fluorescence lifetime to be measured. In the presence of an acceptor when FRET occurs, the donor’s lifetime becomes shorter. As its lifetime is independent of the fluorophore concentration, the FLIM-FRET method allows accurate measurements despite concentration changes.
FRET – the principle
Förster resonance energy transfer (FRET) describes the non-radiative transfer of energy from an excited fluorophore (the donor) to a non-excited different fluorescent molecule (the acceptor) in its vicinity. For a FRET experiment, potentially binding macromolecules, like proteins and nucleic acids, are labeled with spectrally distinct fluorophores in such a way that the emission spectrum of the donor molecule overlaps with the excitation spectrum of the acceptor molecule. Three conditions must be fulfilled for FRET to take place:
- Overlap of donor emission spectrum with acceptor excitation spectrum (Figure 1);
- Donor and acceptor molecules must be in close proximity, less than 10 nanometers (10–8 m), (Figures 2–4); and
- The molecules must have the appropriate relative orientation.
The efficiency (E) of the FRET process depends greatly on the distance between the acceptor and donor, r, where E = 1/[1+ (r/R0)6] and R0 is the Förster radius (refer to Figure 4). If the 3 conditions above are met, the excited donor can transfer its energy to the acceptor. In turn, the acceptor emits a photon and the fluorescence lifetime of the donor molecule decreases.
 must overlap with the excitation spectrum of the acceptor (here EYFP, yellow line).")
 is separated by a distance r from the acceptor molecule (A).")

 is transferred non-radiatively to the acceptor. In turn, the latter emits a photon (yellow arrow). The efficiency (E) of this process is strongly distance dependent.")
The impact
Due to its strong dependence on the distance between the donor and acceptor molecule (r–6, Figure 4), FRET occurs on a spatial scale which is highly relevant for biochemical reactions, such as protein-protein or protein-DNA interactions. FRET can probe molecular interactions by a sensitive fluorescence read-out. This allows researchers to study molecular interactions both in vitro and in vivo. By linking two interaction partners of interest with suitable fluorescent labels, it is possible to analyze bi-molecular interactions. Alternatively, FRET allows the construction of biological probes reporting concentrations of second messengers or ion strength by means of an intra-molecular FRET due to strong conformational change.
Not surprisingly, FRET has developed into a tool widely used in cell biology, biophysics, and biomedical imaging.
The methodology
There are different techniques to detect FRET in the context of microscopy. Commonly known are techniques based on fluorescence intensity of either the donor (acceptor-photobleaching, FRET AB) or the acceptor (sensitized emission, FRET SE).
Intensity-based FRET can be readily applied using standard confocal microscopes. However, it also has some drawbacks. FRET AB cannot be applied in time-series experiments and is susceptible to reversible photobleaching or photoconversion of the donor molecules. FRET SE, on the other hand, suffers from spectral cross talk inherent to all FRET pairs and requires careful calibration measurements as well as linear unmixing of resulting images. A different approach for measuring FRET, which is based on fluorescence lifetime imaging microscopy (FLIM), is described below.
Fluorescence lifetime
The process of fluorescence is often understood in terms of energy transitions from the electronic ground state (S0) to its excited state (S1) in a molecule (Figure 5, left). Such transitions can be elicited by incident light with the appropriate energy, i.e., wavelength. The absorbed energy is stored by the fluorescent molecule for a short time before it can be emitted as fluorescence. The time a molecule spends in its excited state is known as the fluorescence lifetime [1]. It is typically on the order of nanoseconds (10–9 s) for many organic dyes and fluorescent proteins (FPs).
Fluorescence lifetime and FRET
An alternative process to relax from the excited state is, for example, FRET. By FRET excitation energy is non-radiatively transferred to an acceptor molecule. The acceptor in turn can relax by fluorescence (Figure 5, right). Because donor fluorescence and energy transfer are competing processes, the rate depleting the excited state increases in the presence of FRET. One might say, the longer the donor molecules spend in the excited state the more likely it is that FRET occurs. Only those photons from donor molecules which relax by fluorescence are observed. Energy transferred to acceptor molecules is not detected due to the longer wavelength of acceptor fluorescence. Therefore, FRET shortens the donor lifetime (Figure 6).
.")

Figure 6: Plotting the fluorescence photon number over elapsed time after excitation. The initial number of emitted photons after the excitation pulse, a0, decays exponentially. The fluorescence takes time to decay to a0/e (~37%) which is the fluorescence lifetime. Lifetime τ shifts to shorter times due to FRET (τquench). Another read-out from the lifetime decay is the amplitude a0. Measuring the lifetime at each position in a scanning system yields a spatial map of lifetimes (see inset).
Fluorescence lifetime imaging microscopy (FLIM)
Fluorescence lifetimes can be measured in the time domain using pulsed lasers and single photon counting detectors [1,2]. With this approach, the lifetime is determined by building up a histogram of detected fluorescence events. This reveals a single or multi-exponential fluorescence decay. Numerical curve fitting renders the fluorescence lifetime and the amplitude, i.e., the number of detected photons.
FLIM-FRET
Because FRET decreases the donor lifetime, one can quantify the extent to which FRET occurs, provided the donor lifetime without FRET is known. This donor lifetime, τ, serves as an absolute reference against which the FRET sample is analyzed. Therefore, FRET-FLIM is self-calibrated - a property alleviating many of the shortcomings of intensity-based FRET measurements. Because fluorescence lifetime is an inherent property of each dye, it is widely invariant to otherwise detrimental effects, such as photobleaching, image shading, varying concentrations, or expression levels.
The major limitation using intensity-based FRET measurement is the underlying assumption that all observable donor molecules undergo FRET, however, this is usually not the case. This varying "unbound" fraction of donor molecules introduces considerable uncertainty to the measured FRET efficiency, making comparisons between experiments impossible. FRET-FLIM overcomes this disadvantage.
As mentioned previously, the FRET efficiency is computed from the ratio of the fretting donor lifetime, τquench, over the non-fretting lifetime, τ, as:
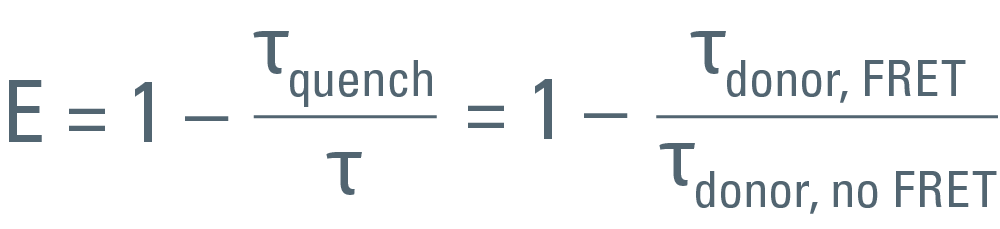
To achieve accuracy, τ must be known from a measurement using a sample which contains only the donor. It is important to exclude any emission from the acceptor. The same settings must be used for both the donor-only measurement as well as the measurement using the FRET sample.
Solution for FLIM-FRET
Perform FLIM-FRET measurements efficiently with the STELLARIS 8 FALCON system . It offers an integrated FRET analysis . TauInteraction is the TauSense tool in the STELLARIS platform that provides the minimal fraction of interacting donors while doing FRET experiments [3].