Light-emitting processes
Some very common biological or biochemical lab methods are based on the existence of several "…escences" such as phosphorescence, chemiluminescence, bioluminescence and finally fluorescence. As an introduction to the topic of Fluorescent Proteins it may be useful to learn a bit more about "…escences". The origin of these phenomena lies in the Latin word for see: -escentia, which already implies the connection to our visual system. All the "…escences" describe physical, chemical or biological processes that we can perceive with our eyes. We add the suffix "…escence" to words denoting change, action or process like the word convalescence.
So obviously, fluorescence is something we can see and something that concerns a change or a process. We will learn how fluorescence fulfills these conditions later on. First we will take a short look at the other "…escences" which are all luminescences themselves. Luminescence is the generic term for the emission of light which is not an effect of high temperature. So luminescence can be determined as an appearance of cold body radiation. This radiation can either be part of a chemical reaction or a cause of subatomic motions or stress on a crystal. Another way to generate emission is incandescence where light is emitted by a substance as the result of heat (e.g. hot metal).
Chemiluminescence is a light-emitting process based on a chemical reaction where the product has an excited intermediate. This intermediate emits light when falling into the ground state. Unlike fluorescence, electrons in chemiluminescent materials are excited by a chemical reaction and not by the absorption of photons. Chemiluminescence finds its technical application in light sticks for example. A well known chemiluminescent substance is luminol, which is used in criminalistics to find blood traces. Here Fe2+ ions which are present in hemoglobin function as a catalyzer to bring Luminol to its light emitting configuration.
If a living organism emits light we speak of bioluminescence, no matter how this light is produced. There are a lot of organisms that produce light, like glowworms (Lampyris noctiluca) or fireflies (Photinus pyralis). A very extraordinary organism in a row of several other fungi is the Jack O'Lantern mushroom (Omphalotus nidiformis), which glows in the dark. Numerous marine organisms like some corals, algae, crustacaea or even squids emit light, mostly in the blue or green spectrum. Another sea inhabitant is the bioluminating jellyfish Aequorea victoria, the source of the green fluorescent protein (GFP). Whereas the firefly, for example uses only a chemiluminescent process to produce light, A. victoria uses both a chemiluminescent and a fluorescent process. As scientists found out, the jellyfish generates blue light by a chemical reaction with the help of the protein Aequorin. This blue light is then used to excite the already mentioned GFP in a fluorescence reaction resulting in a green glow.

This leads us to the question: "What is fluorescence?" Fluorescence is a process where a substance emits light as an effect of the absorption of light of a shorter wavelength. The difference in wavelengths is called Stoke’s Shift. In detail, fluorescence occurs if a substance absorbs light in the form of photons. This leads to a shift of electrons to a higher energy level. But this high energy situation is very unstable, which is why electrons tend to return to their ground state. During this procedure energy is released again in the form of photons that can be seen as a glow. In contrast to phosphorescence, electron energy shift is very fast, in fact in the range of nano seconds (s. Figure 1). Any substance which is able to emit light of a distinct wavelength after excitation with another distinct wavelength is called a fluorochrome. These are discussed in the next section. Most fluorescing substances occurring in nature have a broad excitation and emission spectrum, but substances with clearly defined excitation and emission maxima are more useful for fluorescence microscopy.
Similar to fluorescence, phosphorescence is a light-emitting phenomenon where the phosphorescent material is excited with light. Even though it is closely related to fluorescence, it is much slower. In contrast to fluorescence the re-emission of photons is decelerated by the association of excited electrons energy with a "forbidden" state. Their return to the ground state does not occur as fast as in the case of fluorescence because energy is "trapped". Typical examples of phosphorescent materials are "glow-in-the-dark" toys which can be “charged” with an ordinary light bulb or daylight and then emit light for several minutes or even hours.
Fluorophore
As mentioned above, a fluorochrome is any substance that emits fluorescence. A fluorescent protein is a fluorochrome. Before going into detail, we should introduce several terms and expressions for fluorescent proteins. The term related to fluorescence and more commonly used is fluorophore: a fluorophore is the part of a molecule (such as a protein) that is responsible for producing fluorescence. Thus, GFP or its derivatives are fluorophores of any hybrid protein linked to GFP or its derivatives (e.g., α-microtubulin-GFP). However, a fluorophore does not have to be a protein. Small molecules like FITC, TRITC (20 atoms), or quantum dots (100-100,000 atoms) are also fluorophores.
Looking a little bit deeper into a fluorophore we reach the chromophore, which is the part of a molecule that defines its color (s. Figure 2). Inside the chromophore, energy level transformation of electrons and the interconnected emission of light (see above) take place. There are at least two forms of chromophore. Either they are built of a conjugated π-electron resonance system (GFP) or of metal complexes (chlorophyll, heme).

Because of its relevance for microscopy, we will take a closer look at the chromophore of GFP. It turns out that for the formation of the GFP chromophore no other cofactors or enzyme components are necessary than molecular oxygen. Defined by the primary structure of the GFP amino acid backbone, it forms spontaneously in a self-catalyzed folding mechanism and is completed by intramolecular rearrangements. Going into detail, the chromophore is made of three relevant amino acids. Ser65, Tyr66 and Gly67 undergo a cyclization, a dehydration and an oxidation successively. The result of these reactions is a conjugated π-electron resonance system, the mature GFP chromophore.
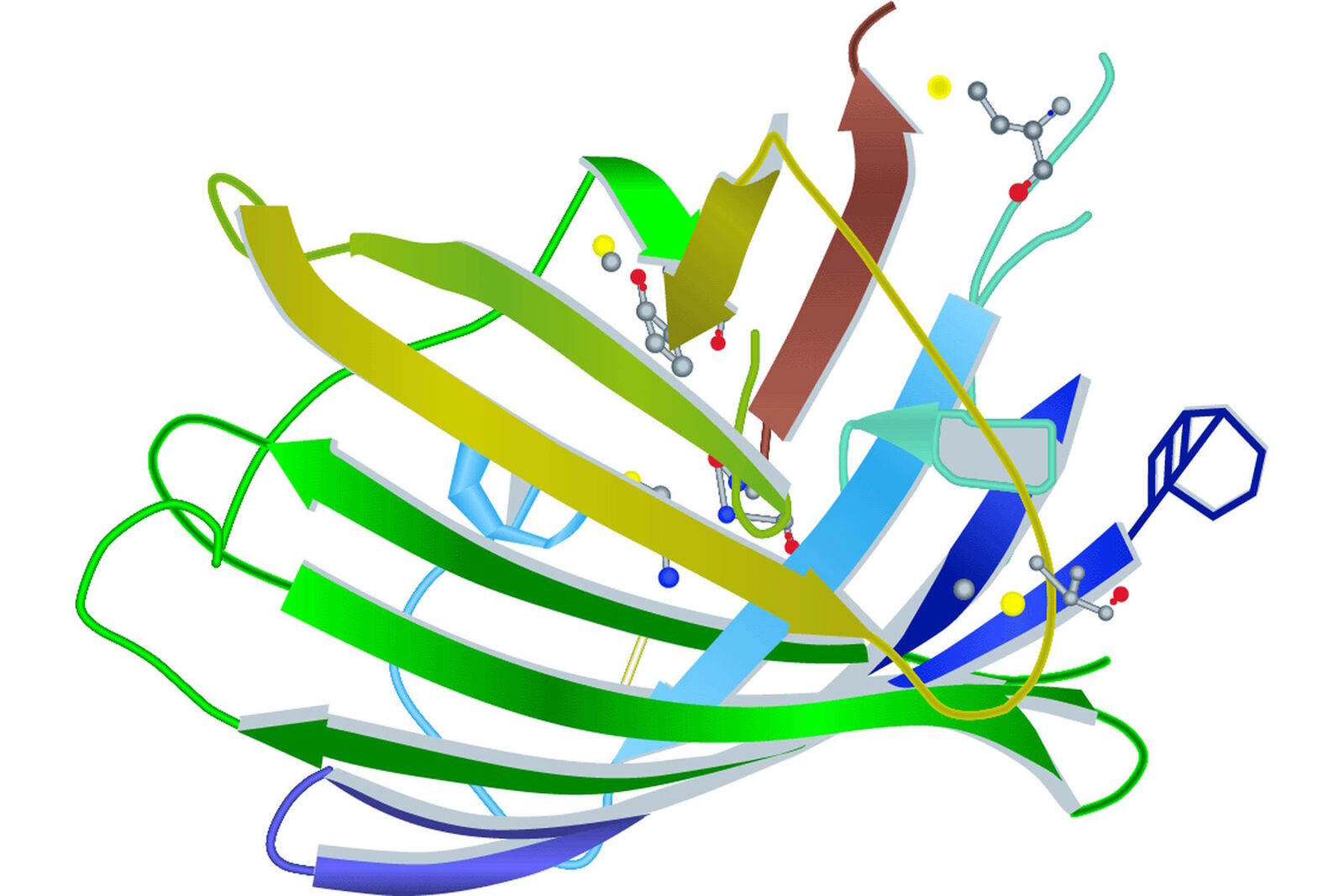
Looking at the entire GFP structure, the cyclic tripeptide sits in the middle of a cylinder. This cylinder is made of 11 strands forming a β-barrel structure, which gives the protein a high stability. The β-barrel structure has a diameter of about 3 nm and a height of about 4 nm (s. Figure 3). All of the FPs known so far have this protecting cylinder which has a great influence on their photophysical properties. In the case of GFP, quantum yield and photostability are relatively high. Furthermore the very compact protein structure leads to a high pH resistance. Specimens with a GFP-tag are comparatively robust concerning temperature and show high tolerance to denaturating substances like paraformaldehyde, urea or guanidinium hydrochloride.
Quenching and bleaching
Nevertheless, there are certain limitations and restrictions for FPs which should be mentioned in the next section. The process of quenching, for example, decreases the intensity of a fluorophore in a reversible way. There are various reasons for this decay. On the one hand there may be a complex formation or an internal conversion. On the other hand quenching can be an effect of an energy transfer process. A microscopic application called FRET takes advantage of this procedure. During Förster-Resonance-Energy-Transfer or Fluorescence-Resonance-Energy-Transfer the energy of a donor fluorophore D is transferred to an acceptor fluorophore A. The fluorescence of D decreases whereas the fluorescence of A increases.
In contrast to quenching which is a reversible process, there is an irreversible fluorescence decrease process called bleaching. Permanent fluorescence loss is based on the destruction of the fluorophore by prolonged exposure to the excitation light. The amount of bleaching depends on the intensity, exposure time and the energy of the light source. Each fluorophore has a certain amount of excitation and emission cycles before it bleaches. In the case of GFP each molecule can be excited about 104–105 times. This complies with 0.1–1 s.
Using a confocal laser scanning microscope light intensities of around 106 W/m2 are common. This high energy dose delivers photons in excess what leads to the phenomenon that a lot of fluorescent molecules are in their excited state. In this case, they no longer absorb light at their usual wavelength. With it, the effective dye concentration is diminished or in other words a pixel which is depicted on the monitor is no longer a direct function of dye concentration. In contrast to illumination with a simple arc lamp, using high energy laser light can lead to a non-linear relation between emission and excitation.
Quantum yield
Talking about the efficiency of fluorescent proteins, we should mention the quantum yield. This is another characteristic of a fluorophore, comparing photon input and output. For the quantum yield calculation of a certain fluorophore, emitted photons are divided by the absorbed ones:
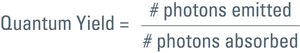
So for a 100 % yield, the quantum yield would be 1. Each photon used for excitation would spawn one photon in the emission. However, this is only a theoretical value. In practice more photons have to be spent for excitation than you get in the emitted light. Quantum yield for GFP, for example is 0.6 (see Table 1). In other words, to get 6 photons in the emission one has to use 10 for the excitation.
Brightness
The brightness of a fluorescent protein is another important characteristic to consider when applying fluorescence microscopy. Depending on the quality of the microscope this parameter is a critical point in the success of an experiment. An objective way to specify the brightness of a fluorescent protein is the multiplication of its molar extinction coefficient with its quantum yield divided by 1,000. The molar extinction coefficient of a substance tells us something about its absorption of light at a distinct wavelength. In detail, the molar extinction coefficient is a dimension for the absorbance of electromagnetic radiation by a substance with molar concentration over a distance of 1 cm at a distinct wavelength. Therefore the corresponding unit is M–1 cm–1.

With the help of this calculation it is possible to compare brightness of different fluorescent proteins. If we take the example of EGFP with a molar extinction coefficient of 56,000 M–1cm–1 and a quantum yield of 0.6, we get a value of 33.6 M–1cm–1. Sometimes people talk about relative brightness of fluorescent proteins. In this case the brightness is calculated by multiplying the molar extinction coefficient with quantum yield and dividing by the brightness value of EGFP (33.6 M–1cm–1). By doing this, relative brightness is converted to well known EGFP which enables slightly faster comparison of different fluorescent proteins.
Photostability
A lot of fluorescent microscopic experiments deal with the observation of living cells. Taking characteristics like quenching and bleaching into consideration, it is obvious that life cell imaging is highly dependent on time. Even in the case of histological or fixed cell samples it is important not to irradiate your specimen for too long a time and too much light intensity. A parameter describing the ability of the fluorescent protein of interest to resist mechanisms degrading its fluorescent behavior is photostability. Photostability is expressed as the number of seconds that pass, until 50 % of the initial brightness has vanished. For comparison, illumination is provided by an arc lamp with a maximum of 10 W/cm2, while a high intensity light source like a laser (up to 100 W/cm2) might produce non-linear effects. Furthermore to measure and compare photostability, pH of fluorescent protein samples is standardized to 7.0 and droplets of protein solutions are adapted to the size of a typical mammalian cell. Adjacent illumination with the above mentioned light source is continuously. In parallel the emission of photons is counted. In the case of EGFP it takes 174 sec for half of the original brightness to fade (Shaner et al. 2005). In other words, emission decrease from 1,000 to 500 photons/sec takes 174 sec.
Knowing different characteristics like excitation and emission maxima, brightness, quantum yield etc. of a certain fluorescent dye or protein the experimenter is able to choose a distinct candidate or observation condition to fulfill his requirements for a certain experimental setup. Furthermore he is capable of interpreting results in a detailed way if he is aware of photo physical features of different fluorescent dyes or proteins.
Read more about fluorescent proteins, their photo spectral characteristics, and their applications
References
Header photo: © by David Mülheims, Germany
 and phalloidin (magenta), imaged using Viventis SCAPE; scale bar 50μm. Courtesy of Marina Cuenca and Heleen Jungen (Dayton lab), EMBL Barcelona.")
, tubulin with Cy5 (red), and nuclei with DAPI (blue). Image courtesy of Dr. Günter Giese, Max Planck Institute for Medical Research, Heidelberg, Germany.")
, Tropomyosin (cardiomyocytes, red) and GFP (primordial cardiac layer, green).")
