Example: Four-Color Time-Lapse Imaging – Confocal
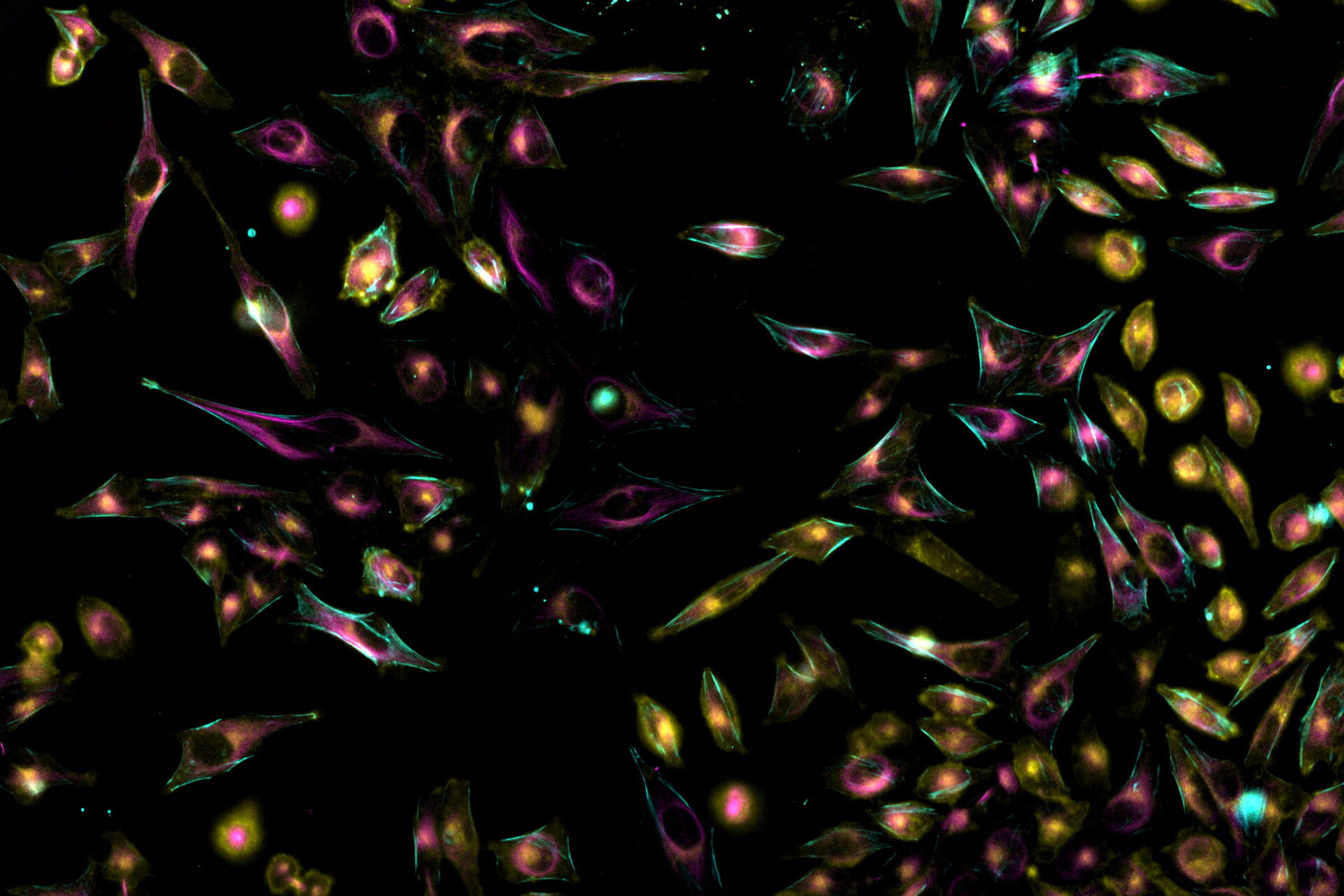
Here is one example showing simultaneous imaging in confocal mode.
, SPY-Actin (cyan), and SiR-Tubulin (magenta). Instant Computational Clearing (ICC) was applied.")
In this article, we provide examples about the ability of Mica to do dynamic live-cell imaging. Live-cell imaging sheds light on diverse cellular events. As many of these events have fast dynamics, the microscope imaging system must be fast enough to record every detail. One major advantage of such an imaging system would be the ability to capture several fluorescence imaging channels simultaneously for their exact spatiotemporal correlation. Mica enables users to acquire the signal of up to four fluorophores at a time, either in confocal or widefield mode. With it, cellular components can be imaged at the very same point in time, without spatiotemporal mismatch.
Here is one example showing simultaneous imaging in confocal mode.
 and phalloidin (magenta), imaged using Viventis SCAPE; scale bar 50μm. Courtesy of Marina Cuenca and Heleen Jungen (Dayton lab), EMBL Barcelona.")
Organoids and other complex in vitro models (CIVMs) are becoming increasingly important in early…

Spatial Proteomics, Nature Methods 2024 Method of the Year, is driving research advancements in…

This article provides an introduction to the recommendations of 21 CFR Part 11 from the FDA,…
