
About Prof. Dr. Jacco van Rheenen

Jacco van Rheenen was originally trained in a variety of imaging techniques during his PhD with Dr. Kees Jalink at the Netherlands Cancer Institute. He was among the first to optimize imaging and develop software to quantitatively measure FRET on confocal microscopes. During his PhD in the lab of Dr. Jalink and as postdoc in the lab of Dr. Sonnenberg (Netherlands Cancer Institute) he used several microscopy techniques to study lipid signaling in tumor cells. In order to broaden his scales, he obtained a KWF fellowship to do a postdoc in the United States in the lab of Dr. John Condeelis. There he extended his imaging experience by imaging mammary tumors intravitally including two-photon microscopy and became an expert in the field of intravital FRET imaging. In 2008 he was appointed as group leader at the Hubrecht Institute, where he utilizes his imaging techniques to visualize processes that are required for the metastasis of mammary tumor cells in living animals. In July 2014 he was appointed professor in Intravital Microscopy at the University Medical Center Utrecht. In 2009, he was awarded a VIDI award from Netherlands Organisation for Scientific Research. In 2013, he received the Stem Cells Young Investigator Award In 2015, he was awarded an ERC consolidator grant, and in 2017 the Dr. Josef Steiner Cancer Research Foundation Award 2017.
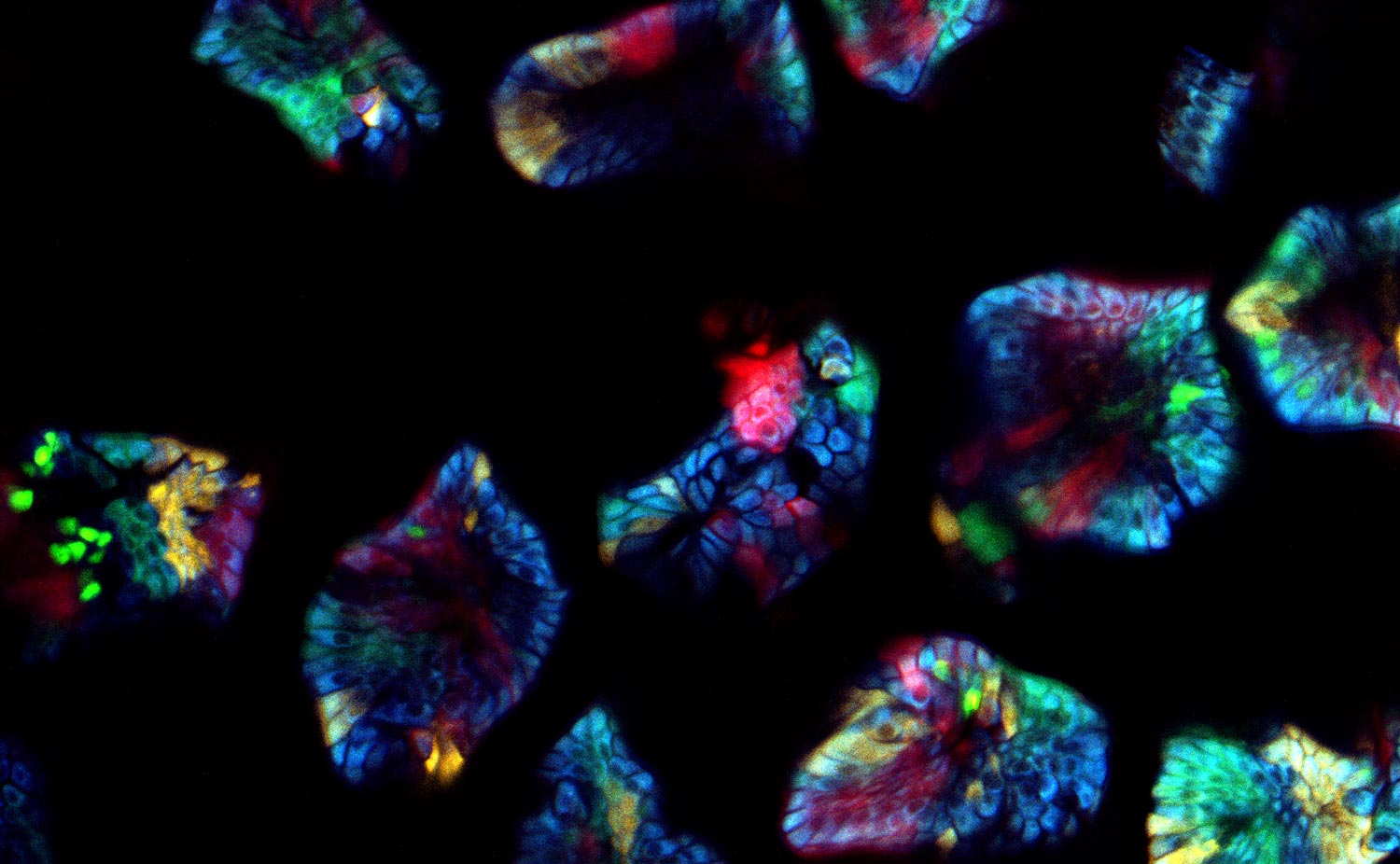
What challenges do you face when imaging confetti mice samples?
Prof. Dr. van Rheenen: There are three main challenges of imaging confetti mice. It is always challenging to image deep into mice and still obtain images with great contrast. For great contrast, you want to detect every photon, but unfortunately lots of light gets scattered in tissues and is therefore lost.
The other challenge is to avoid image distortions caused by respiration movements. One way of avoiding these distortions is by imaging the tissues very fast. However, this also leads to less light collection and therefore less imaging contrast.
The last main challenge is more confetti specific, and is the ability to image and distinguish all the four confetti colors. To do so, the fluorophores can be excited with a single optimized wavelength in sequential mode, but this leads to slow imaging acquisition times and therefore to potential imaging distortions. To avoid this, we use a single sub-optimal wavelength that excite all fluorophores simultaneously, and in that case the fluorophores should be solely distinguished based on their emission spectrums. Unfortunately, the emission spectrums of the confetti fluorophores overlap extensively. The conventional dichroics are never optimal to distinguish all fluorophores, but with the spectral detectors [of the SP8 DIVE] this is now possible and much easier, since we can really optimize the wavelength for each fluorophore you want to detect.
What about the difficulties experienced when imaging such samples with conventional confocal MP instrumentation?
Prof. Dr. van Rheenen: The cells in our confetti sample can either express CFP, GFP, YFP or RFP, and you want to image them all and fast. This leads to serious problems. First, all these dyes cannot be optimally excited with a single wavelength. One can use sequential imaging with different wavelength but that doubles the acquisition time and therefore the chance of image distortion caused by respiration movements. Moreover, due to the chromatic aberration of the different excitation wavelength, the different confetti images will not be acquired at the same z-level. That induces also artifacts.
What’s the big advantage of spectral NDD imaging for tomorrow’s samples?
Prof. Dr. van Rheenen: There is a huge development with new probes and every probe has its own optimized excitation and emission spectrum. Especially for the emission spectrum it’s tedious: You can change one or two filters, but you are not very flexible. When you are in a sample that has not one fluorophore but two fluorophores the system has to be optimized all the time. And you don’t have all the filters. With the spectral detector part it’s much more flexible, you optimize the detection of each fluorophore. Since you are fighting for every photon that’s coming out of the fluorophore, you really want to optimize what part of the spectrum you want to detect. Moreover, we often stitch hundreds of Z-stacks in order to image with high resolution large areas of tissues. By optimizing the detection wavelengths, we can image multiple fluorophores simultaneously, leading to fast imaging acquisitions, which enables us to image large tile scans over time.
Are there things you would envision that are possible today?
Prof. Dr. van Rheenen: Yes, for example, we couldn’t really distinguish between autofluorescence and for example YFP. Although their emission spectrums are clearly different, due to their overlap in the yellow-wavelength range, the conventional dichroics cannot distinguish them. With the spectral detectors, we can now acquire true emission spectrums and thereby really separating largely overlapping fluorophores that we could not before.
What scientific impact and outlook could there be from your work on confetti mice imaged with non descanned spectral detection?
Prof. Dr. van Rheenen: For many research questions that many people have, it’s so important to visualize the behavior of cells over hours, days, weeks, and sometimes months. And the problem is that in many tissues, the position of cells, and even the very cellular composition, change dramatically over the time. Therefore it’s absolutely impossible to visualize the same cells over prolonged times.
These confetti mice are absolutely unique because you can label individual cells very sporadically. Therefore you can trace them and image individual cells over long time periods and study how they behave. The spectral separating NDD is great since we now optimally detect all dyes simultaneously. This is important for experiments where one wants to follow multiple cell types or states that are differentially color coded. In our research, we use this to for example to study simultaneously stem cells and more differentiated cells and visualize how they behave differently in the same tissue. And this is really key for many of the biological questions that we address today and tomorrow.
Related Articles
-
 where cyan indicates nuclei and magenta tight junctions.")
Rapid Check of Live Stem Cells in Cell-Culture Inserts set in Multi-Well Plates
See how efficient imaging of live iPSC stem cells within cell-culture inserts set in a multi-well…
Mar 20, 2024Read article -

Notable AI-based Solutions for Phenotypic Drug Screening
Learn about notable optical microscope solutions for phenotypic drug screening using 3D-cell…
Dec 06, 2023Read article -
. Image courtesy of Prof. Hui Guo, School of Life Sciences, Central South University, China")
How Microscopy Helps the Study of Mechanoceptive and Synaptic Pathways
In this podcast, Dr Langenhan explains how microscopy helps his team to study mechanoceptive and…
Jun 22, 2023Read article
